2026
Machine Learning Reveals Hidden Dimensions of Functional Similarity
Noor Youssef, Sarah Gurev, and Debora S. Marks
PNAS Commentary; 2 January 2026
Links
2025
Biological Sequence Kernels with Guaranteed Flexibility
Alan Nawzad Amin, Debora S. Marks, Eli Nathan Weinstein
Journal of Machine Learning Research; 28 December 2025
Abstract
Applying machine learning to biological sequences---DNA, RNA and protein---has enormous potential to advance human health and environmental sustainability. To support such high-stakes applications, it is important to develop models and evaluations that not only capture underlying biology, but also have theoretical guarantees of reliability and performance. In this article, we analyze kernel methods for biological sequences, including both hand-crafted kernels and deep neural network-based kernels. We show that popular biological kernels can severely fail at learning functions or distinguishing distributions. We then develop modified kernels that (1) are universal, characteristic, and metrize the space of distributions, and (2) preserve the underlying biological inductive biases and domain knowledge embedded in the original kernel. Our results rest on novel proof techniques for kernels that handle the structure of biological sequence space--discrete, variable length sequences--and biological notions of sequence similarity. We illustrate our theoretical results in simulation and on real biological data sets.
Links
Proteome-wide model for human disease genetics
Rose Orenbuch, Courtney A. Shearer, Aaron W. Kollasch, Aviv D. Spinner, Thomas Hopf, Lood van Niekerk, Dinko Franceschi, Mafalda Dias, Jonathan Frazer & Debora S. Marks
Nature Genetics; 24 November 2025
Abstract
Missense variants remain a challenge in genetic interpretation owing to their subtle and context-dependent effects. Although current prediction models perform well in known disease genes, their scores are not calibrated across the proteome, limiting generalizability. To address this knowledge gap, we developed popEVE, a deep generative model combining evolutionary and human population data to estimate variant deleteriousness on a proteome-wide scale. popEVE achieves state-of-the-art performance without overestimating the burden of deleterious variants and identifies variants in 442 genes in a severe developmental disorder cohort, including 123 novel candidates. These genes are functionally similar to known disease genes, and their variants often localize to critical regions. Remarkably, popEVE can prioritize likely causal variants using only child exomes, enabling diagnosis even without parental sequencing. This work provides a generalizable framework for rare disease variant interpretation, especially in singleton cases, and demonstrates the utility of calibrated, evolution-informed scoring models for clinical genomics.
Links
Democratizing protein language model training, sharing and collaboration
Jin Su, Zhikai Li, Tianli Tao, Chenchen Han, Yan He, Fengyuan Dai, Qingyan Yuan, Yuan Gao, Tong Si, Xuting Zhang, Yuyang Zhou, Junjie Shan, Xibin Zhou, Xing Chang, Shiyu Jiang, Dacheng Ma, Anthony Gitter, Milot Mirdita, Kevin K. Yang, Pascal Notin, Debora S. Marks, Pranam Chatterjee, Rohit Singh, Philip A. Romero, Michael Heinzinger, Jianming Liu, Jia Zheng, Stan Z. Li, Anping Zeng, Huaizong Shen, Jijie Chai, Feng Ju, Noelia Ferruz, Anum Glasgow, Philip M. Kim, Christopher Snow, Vasilis Ntranos, Jianyi Yang, Liang Hong, Caixia Gao, Tong Si, Michael Bronstein, Xing Chang, Martin Steinegger, Sergey Ovchinnikov, Fajie Yuan, Jin Su, Zhikai Li, Tianli Tao, Chenchen Han, Yan He, Fengyuan Dai, Xuting Zhang, Yuyang Zhou, Junjie Shan, Xibin Zhou, Shiyu Jiang, Dacheng Ma, Yuan Gao, Jiawei Zhang, Yuliang Fan, Yuyang Tao, Linqi Cheng, Xinzhe Zheng, Lei Chen, Rui Long, Lingjie Kong, Zhongji Pu, Jiaming Guan, Tianyuan Zhang, Cheng Li & Qingyan Yuan, Martin Steinegger, Sergey Ovchinnikov & Fajie Yuan
Nature Biotechnology; 24 October 2025
Abstract
Training and deploying large-scale protein language models typically requires deep machine learning expertise—a barrier for researchers outside this field. SaprotHub overcomes this challenge by offering an intuitive platform that facilitates training and prediction as well as storage and sharing of models. Here we provide the ColabSaprot framework built on Google Colab, which potentially powers hundreds of protein training and prediction applications, enabling researchers to collaboratively build and share customized models.
Links
Pancreatic cancer risk prediction using deep sequential modeling of longitudinal diagnostic and medication records
Chunlei Zheng, Asif Khan, Daniel Ritter, Debora S. Marks, Nhan V. Do, Nathanael R. Fillmore, Chris Sander
Cell Reports Medicine; 16 September 2025
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a rare, aggressive cancer often diagnosed late with low survival rates, due to the lack of population-wide screening programs and the high cost of early detection methods. To enable early detection of high-risk individuals, we develop a transformer-based model trained on longitudinal Veterans Affairs electronic health record (EHR) with 19,426 PDAC cases and ∼15.9 million controls. Our model combines diagnostic and medication trajectories to predict PDAC risk within a 6-, 12-, and 36-month assessment window. Incorporating medication significantly improved performance; among the top 1,000–5,000 highest-risk patients in a cohort of 1 million patients, 3-year PDAC incidence is 115–70 times higher than a reference estimate based on age and sex alone. Furthermore, analysis of most predictive features highlights the role of events such as chronic inflammatory conditions and specific medications on overall PDAC risk. Our work provides an AI-driven identification of high-risk individuals, with a potential to improve early detection, enhance patient care, and reduce healthcare costs.
Links
Results of the Protein Engineering Tournament: An Open Science Benchmark for Protein Modeling and Design
Chase Armerm Hassan Kane, Dana L. Cortade, Henning Redestig, David A. Estell, Adil Yusuf, Nathan Rollins, Aviv Sprinner, Debora Marks, TJ Brunette, Peter J. Kelly, Erika DeBenedictis
PROTEINS; 23 June 2025
Abstract
The grand challenge of protein engineering is the development of computational models to characterize and generate protein sequences for arbitrary functions. Progress is limited by lack of 1) benchmarking opportunities, 2) large protein function datasets, and 3) access to experimental protein characterization. We introduce the Protein Engineering Tournament—a fully-remote competition designed to foster the development and evaluation of computational approaches in protein engineering. The tournament consists of an in silico round, predicting biophysical properties from protein sequences, followed by an in vitro round where novel protein sequences are designed, expressed and characterized using automated methods. Upon completion, all datasets, experimental protocols, and methods are made publicly available. We detail the structure and outcomes of a pilot Tournament involving seven protein design teams, powered by six multi-objective datasets, with experimental characterization by our partner, International Flavors and Fragrances. Forthcoming Protein Engineering Tournaments aim to mobilize the scientific community towards transparent evaluation of progress in the field.
Links
Evolutionary-Scale Enzymology Enables Biochemical Constant Prediction Across a Multi-Peaked Catalytic Landscape
Duncan F. Muir, Garrison P. R. Asper, Pascal Notin, Jacob A. Posner, Debora S. Marks, Michael J. Keiser, Margaux M. Pinney
pre-print
Abstract
Quantitatively mapping enzyme sequence-catalysis landscapes remains a critical challenge in understanding enzyme function, evolution, and design. Here, we expand an emerging microfluidic platform to measure catalytic constants—kcat and KM—for hundreds of diverse naturally occurring sequences and mutants of the model enzyme Adenylate Kinase (ADK). This enables us to dissect the sequence-catalysis landscape’s topology, navigability, and mechanistic underpinnings, revealing distinct catalytic peaks organized by structural motifs. These results challenge long-standing hypotheses in enzyme adaptation, demonstrating that thermophilic enzymes are not slower than their mesophilic counterparts. Combining the rich representations of protein sequences provided by deep-learning models with our custom high-throughput kinetic data yields semi-supervised models that significantly outperform existing models at predicting catalytic parameters of naturally occurring ADK sequences. Our work demonstrates a promising strategy for dissecting sequence-catalysis landscapes across enzymatic evolution and building family-specific models capable of accurately predicting catalytic constants, opening new avenues for enzyme engineering and functional prediction.
Links
Protein design for evaluating vaccines against future viral variation
Noor Youssef, Sarah Gurev, Fadi Ghantous, Kelly Brock, Javier Jaimes, Nicole N. Thadani, Ann Dauphin, Amy C. Sherman, Leonid Yurkovetskiy, Daria Soto, Ralph Estanboulieh, Ben Kotzen, Pascal Notin, Aaron W. Kollasch, Alexander A. Cohen, Sandra E. Dross, Jesse H. Erasmus, Deborah H. Fuller, Pamela J. Bjorkman, Jacob E. Lemieux, Jeremy Luban, Michael S. Seaman, Debora S. Marks
Immunity, 8 May 2025
Summary
Recurrent waves of viral infection necessitate vaccines and therapeutics that remain effective against emerging viruses. Our ability to evaluate interventions is currently limited to assessments against past or circulating variants, which likely differ in their immune escape potential compared with future variants. To address this, we developed EVE-Vax, a computational method for designing antigens that foreshadow immune escape observed in future viral variants. We designed 83 SARS-CoV-2 spike proteins that transduced ACE2-positive cells and displayed neutralization resistance comparable to variants that emerged up to 12 months later in the COVID-19 pandemic. Designed spikes foretold antibody escape from B.1-BA.4/5 bivalent booster sera seen in later variants. The designed constructs also highlighted the increased neutralization breadth elicited by nanoparticle-based, compared with mRNA-based, boosters in non-human primates. Our approach offers targeted panels of synthetic proteins that map the immune landscape for early vaccine and therapeutic evaluation against future viral strains.
Links
Machine learning prediction of enzyme optimum pH
Japheth E. Gado, Matthew Knotts, Ada Y. Shaw, Debora S. Marks, Nicholas P. Gauthier, Chris Sander & Gregg T. Beckham
Nature Machine Intelligence, 29 April 2025
Abstract
The relationship between pH and enzyme catalytic activity, especially the optimal pH (pHopt) at which enzymes function, is critical for biotechnological applications. Hence, computational methods to predict pHopt will enhance enzyme discovery and design by facilitating accurate identification of enzymes that function optimally at specific pH levels, and by elucidating sequence–function relationships. Here we proposed and evaluated various machine learning methods for predicting pHopt, conducting extensive hyperparameter optimization and training over 11,000 model instances. Our results demonstrate that models utilizing language model embeddings markedly outperform other methods in predicting pHopt. We present EpHod, the best-performing model, to predict pHopt, making it publicly available to researchers. From sequence data, EpHod directly learns structural and biophysical features that relate to pHopt, including proximity of residues to the catalytic centre and the accessibility of solvent molecules. Overall, EpHod presents a promising advancement in pHopt prediction and will potentially speed up the development of enzyme technologies.
Links
Custom CRISPR—Cas9 PAM variants via scalable engineering and machine learning
Rachel A. Silverstein, Nahye Kim, Ann-Sophie Kroell, Russell T. Walton, Justin Delano, Rossano M. Butcher, Martin Pacesa, Blaire K. Smith, Kathleen A. Christie, Leillani L. Ha, Ronald J. Meis, Aaron B. Clark, Aviv D. Spinner, Cicera R. Lazzarotto, Yichao Li, Azusa Matsubara, Elizabeth O. Urbina, Gary A. Dahl, Bruno E. Correia, Debora S. Marks, Shengdar Q. Tsai, Luca Pinello, Suk See De Ravin, Qin Liu & Benjamin P. Kleinstiver
Nature, 22 April 2025
Abstract
Engineering and characterizing proteins can be time-consuming and cumbersome, motivating the development of generalist CRISPR-Cas enzymes1–4 to enable diverse genome editing applications. However, such enzymes have caveats such as an increased risk of off-target editing3,5,6. To enable scalable reprogramming of Cas9 enzymes, here we combined high-throughput protein engineering with machine learning (ML) to derive bespoke editors more uniquely suited to specific targets. Via structure/function-informed saturation mutagenesis and bacterial selections, we obtained nearly 1,000 engineered SpCas9 enzymes and characterized their protospacer-adjacent motif7 (PAM) requirements to train a neural network that relates amino acid sequence to PAM specificity. By utilizing the resulting PAM ML algorithm (PAMmla) to predict the PAMs of 64 million SpCas9 enzymes, we identified efficacious and specific enzymes that outperform evolution-based and engineered SpCas9 enzymes as nucleases and base editors in human cells while reducing off-targets. An in silico directed evolution method enables user-directed Cas9 enzyme design, including for allele-selective targeting of the RHO P23H allele in human cells and mice. Together, PAMmla integrates ML and protein engineering to curate a catalog of SpCas9 enzymes with distinct PAM requirements, and motivates the use of efficient and safe bespoke Cas9 enzymes instead of generalist enzymes for various applications.
Links
SEQUENCE-BASED PROTEIN MODELS FOR THE PREDICTION OF MUTATIONS ACROSS PRIORITY VIRUSES
Sarah Gurev, Noor Youssef, Navami Jain, Debora S. Marks
GEM workshop, ICLR 2025
Abstract
Viruses pose a significant threat to human health. Advances in machine learning for predicting mutation effects have enhanced viral surveillance and enabled the proactive design of vaccines and therapeutics, but the accuracy of these methods across priority viruses remain unclear. We perform the first large-scale modeling across 40 WHO priority pandemic-threat pathogens, many of which are undersurveilled, discovering that most have sufficient sequence or structural information for effective modeling, highlighting the potential for using these approaches in pandemic preparedness. To understand the limits of current modeling capabilities for viruses, we curate 51 standardized viral deep mutational scanning assays to systematically evaluate the performance of three alignment-based models, three Protein Language Models (PLMs), and two structure-aware PLMs with different training databases. We find marked differences in performance of these models on viruses relative to non-viral proteins. For viral proteins, we find alignment-based models perform on par with PLMs though with predictable differences in which model is better for a particular function or virus depending on data available. We define confidence metrics for both alignment-based models and PLMs that indicate when additional sequence or structural data may be needed for accurate predictions and to guide model selection in the absence of available data for evaluation. We use these metrics to inform the development a confidence-weighted hybrid model that builds on the strength of each approach, adapts to the quality of data available, and outperforms either of the best alignment or PLM models alone.
Links
Evolutionary-Scale Enzymology Enables Biochemical Constant Prediction Across a Multi-Peaked Catalytic Landscape
Duncan F. Muir, Garrison P. R. Asper, Pascal Notin, Jacob A. Posner, Debora S. Marks, Michael J. Keiser, Margaux M. Pinney
Biorxiv, preprint
Abstract
Quantitatively mapping enzyme sequence-catalysis landscapes remains a critical challenge in understanding enzyme function, evolution, and design. Here, we expand an emerging microfluidic platform to measure catalytic constants—kcat and KM—for hundreds of diverse naturally occurring sequences and mutants of the model enzyme Adenylate Kinase (ADK). This enables us to dissect the sequence-catalysis landscape’s topology, navigability, and mechanistic underpinnings, revealing distinct catalytic peaks organized by structural motifs. These results challenge long-standing hypotheses in enzyme adaptation, demonstrating that thermophilic enzymes are not slower than their mesophilic counterparts. Combining the rich representations of protein sequences provided by deep-learning models with our custom high-throughput kinetic data yields semi-supervised models that significantly outperform existing models at predicting catalytic parameters of naturally occurring ADK sequences. Our work demonstrates a promising strategy for dissecting sequence-catalysis landscapes across enzymatic evolution and building family-specific models capable of accurately predicting catalytic constants, opening new avenues for enzyme engineering and functional prediction.
Links
MaveDB 2024: a curated community database with over seven million variant effects from multiplexed functional assays
Alan F. Rubin, Jeremy Stone, Aisha Haley Bianchi, Benjamin J. Capodanno, Estelle Y. Da, Mafalda Dias, Daniel Esposito, Jonathan Frazer, Yunfan Fu, Sally B. Grindstaff, Matthew R. Harrington, Iris Li, Abbye E. McEwen, Joseph K. Min, Nick Moore, Olivia G. Moscatelli, Jesslyn Ong, Polina V. Polunina, Joshua E. Rollins, Nathan J. Rollins, Ashley E. Snyder, Amy Tam, Matthew J. Wakefield, Shenyi Sunny Ye, Lea M. Starita, Vanessa L. Bryant, Debora S. Marks & Douglas M. Fowler
Genome Biology, Jan 2025
Abstract
Multiplexed assays of variant effect (MAVEs) are a critical tool for researchers and clinicians to understand genetic variants. Here we describe the 2024 update to MaveDB (https://www.mavedb.org/) with four key improvements to the MAVE community’s database of record: more available data including over 7 million variant effect measurements, an improved data model supporting assays such as saturation genome editing, new built-in exploration and visualization tools, and powerful APIs for data federation and streamlined submission and access. Together these changes support MaveDB’s role as a hub for the analysis and dissemination of MAVEs now and into the future.
Links
Multi-megabase scale genome interpretation with genetic language models
Frederik Träuble, Lachlan Stuart, Andreas Georgiou, Pascal Notin, Arash Mehrjou, Ron Schwessinger, Mathieu Chevalley, Kim Branson, Bernhard Schölkopf, Cornelia van Duijn, Debora Marks, Patrick Schwab
Biorxiv, pre-print
Abstract
Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.
Links
2024
Multi-Scale Representation Learning for Protein Fitness Prediction
Zuobai Zhang, Pascal Notin, Yining Huang, Aurélie Lozano, Vijil Chenthamarakshan, Debora Marks, Payel Das, Jian Tang
NeurIPS, 11 December 2024
Abstract
Designing novel functional proteins crucially depends on accurately modeling their fitness landscape. Given the limited availability of functional annotations from wet-lab experiments, previous methods have primarily relied on self-supervised models trained on vast, unlabeled protein sequence or structure datasets. While initial protein representation learning studies solely focused on either sequence or structural features, recent hybrid architectures have sought to merge these modalities to harness their respective strengths. However, these sequence-structure models have so far achieved only incremental improvements when compared to the leading sequence-only approaches, highlighting unresolved challenges effectively leveraging these modalities together. Moreover, the function of certain proteins is highly dependent on the granular aspects of their surface topology, which have been overlooked by prior models. To address these limitations, we introduce the Sequence-Structure-Surface Fitness (S3F) model - a novel multimodal representation learning framework that integrates protein features across several scales. Our approach combines sequence representations from a protein language model with Geometric Vector Perceptron networks encoding protein backbone and detailed surface topology. The proposed method achieves state-of-the-art fitness prediction on the ProteinGym benchmark encompassing 217 substitution deep mutational scanning assays, and provides insights into the determinants of protein function.
Links
Toward trustable use of machine learning models of variant effects in the clinic
Mafalda Dias, Rose Orenbuch, Debora S. Marks, Jonathan Frazer
American Journal of Human Genetics; 18 November 2024
Abstract
There has been considerable progress in building models to predict the effect of missense substitutions in protein-coding genes, fueled in large part by progress in applying deep learning methods to sequence data. These models have the potential to enable clinical variant annotation on a large scale and hence increase the impact of patient sequencing in guiding diagnosis and treatment. To realize this potential, it is essential to provide reliable assessments of model performance, scope of applicability, and robustness. As a response to this need, the ClinGen Sequence Variant Interpretation Working Group, Pejaver et al., recently proposed a strategy for validation and calibration of in-silico predictions in the context of guidelines for variant annotation. While this work marks an important step forward, the strategy presented still has important limitations. We propose core principles and recommendations to overcome these limitations that can enable both more reliable and more impactful use of variant effect prediction models in the future.
Links
Continuous evolution of user-defined genes at 1 million times the genomic mutation rate
Gordon Rix, Rory Williams, Vincent Hu, Aviv Spinner, Alexander (Olek) Pisera, Debora Marks, Chang Liu
Science; 8 November 2024
Abstract
When a gene evolves under prevailing and shifting functional demands, those demands become embedded into the resulting diversity of homologous sequences as patterns of conservation and change. We have long used these patterns to extract features important for gene function and to infer how genes historically changed to satisfy new demands, offering lessons in biomolecular design. Yet only natural evolution, through millions of years of operation, has consistently diverged genes far enough to yield sets of homologous sequences containing abundant statistical information on the details of their function. Compressing long-term gene evolution to laboratory time spans at scale would break nature’s monopoly on the production of extensive gene diversity and would enable the systematic detection of additional structural and functional demands governing biology, the engineering of custom biomolecules, and the prospective study of evolutionary mechanisms and principles by which gene diversity was generated in the first place.
Links
An ANXA11 P93S variant dysregulates TDP-43 and causes corticobasal syndrome
Allison Snyder, Veronica H Ryan, James Hawrot, Sydney Lawton, Daniel M Ramos, Y Andy Qi, Kory Johnson, Xylena Reed, Nicholas L Johnson, Aaron W Kollasch, Megan Duffy, Lawren VandeVrede, J Nicholas Cochran, Bruce L Miller, Camilo Toro, Bibiana Bielekova, Jennifer S Yokoyama, Debora S Marks, Justin Y Kwan, Mark R Cookson, Michael E Ward
Alzheimer’s & Dementia; 26 June 2024
Abstract
As genetic testing has become more accessible and affordable, variants of uncertain significance (VUS) are increasingly identified, and determining whether these variants play causal roles in disease is a major challenge. The known disease-associated Annexin A11 (ANXA11) mutations result in ANXA11 aggregation, alterations in lysosomal-RNA granule co-trafficking, and TDP-43 mis-localization and present as amyotrophic lateral sclerosis or frontotemporal dementia. We identified a novel VUS in ANXA11 (P93S) in a kindred with corticobasal syndrome and unique radiographic features that segregated with disease. We then queried neurodegenerative disorder clinic databases to identify the phenotypic spread of ANXA11 mutations. Multi-modal computational analysis of this variant was performed and the effect of this VUS on ANXA11 function and TDP-43 biology was characterized in iPSC-derived neurons. Single-cell sequencing and proteomic analysis of iPSC-derived neurons and microglia were used to determine the multiomic signature of this VUS. Mutations in ANXA11 were found in association with clinically diagnosed corticobasal syndrome, thereby establishing corticobasal syndrome as part of ANXA11 clinical spectrum. In iPSC-derived neurons expressing mutant ANXA11, we found decreased colocalization of lysosomes and decreased neuritic RNA as well as decreased nuclear TDP-43 and increased formation of cryptic exons compared to controls. Multiomic assessment of the P93S variant in iPSC-derived neurons and microglia indicates that the pathogenic omic signature in neurons is modest compared to microglia. Additionally, omic studies reveal that immune dysregulation and interferon signaling pathways in microglia are central to disease. Collectively, these findings identify a new pathogenic variant in ANXA11, expand the range of clinical syndromes caused by ANXA11 mutations, and implicate both neuronal and microglia dysfunction in ANXA11 pathophysiology. This work illustrates the potential for iPSC-derived cellular models to revolutionize the variant annotation process and provides a generalizable approach to determining causality of novel variants across genes.
Links
FUTURE-PROOF VACCINE DESIGN WITH A GENERATIVE MODEL OF ANTIBODY CROSS-REACTIVITY
Sarah Gurev, Noor Youssef, Hannah Pierce-Hoffman, Debora S. Marks
GEM workshop, ICLR 2024
Abstract
Mosaic nanoparticle vaccines incorporating naturally diverse sarbecovirus receptor binding domains (RBDs) represent a promising approach for pan-coronavirus vaccine design. Mosaic nanoparticles elicit broad, cross-reactive immune responses, likely because antibodies utilize avidity effects to preferentially bind to conserved regions where they can cross-link across neighboring RBDs. However, the diversity in natural RBDs is limited, leading to ‘off-target’ antibodies that do not bind to low-mutability regions. We therefore develop a novel futureproof vaccine design method, building upon a probabilistic generative model of antibody escape, to computationally design RBDs with further diversity. This approach aims to focus antibody responses to regions that are (1) neutralizing, (2) accessible and (3) unlikely to mutate during future viral evolution. The designs will be assessed by immunizing mice and testing the breadth of neutralizability of the sera compared to a nanoparticle composed of naturally diverse strains.
Links
FUTURE-PROOF VACCINE DESIGN WITH A GENERATIVE MODEL OF ANTIBODY CROSS-REACTIVITY
Sarah Gurev, Noor Youssef, Hannah Pierce-Hoffman, Debora S. Marks
GEM workshop, ICLR 2024; 11 May 2024
Abstract
Mosaic nanoparticle vaccines incorporating naturally diverse sarbecovirus receptor binding domains (RBDs) represent a promising approach for pan-coronavirus vaccine design. Mosaic nanoparticles elicit broad, cross-reactive immune responses, likely because antibodies utilize avidity effects to preferentially bind to conserved regions where they can cross-link across neighboring RBDs. However, the diversity in natural RBDs is limited, leading to ‘off-target’ antibodies that do not bind to low-mutability regions. We therefore develop a novel futureproof vaccine design method, building upon a probabilistic generative model of antibody escape, to computationally design RBDs with further diversity. This approach aims to focus antibody responses to regions that are (1) neutralizing, (2) accessible and (3) unlikely to mutate during future viral evolution. The designs will be assessed by immunizing mice and testing the breadth of neutralizability of the sera compared to a nanoparticle composed of naturally diverse strains.
Links
SaprotHub: Making Protein Modeling Accessible to All Biologists
Jin Su, Zhikai Li, Chenchen Han, Yuyang Zhou, Yan He, Junjie Shan, Xibin Zhou, Xing Chang, Shiyu Jiang, Dacheng Ma, The OPMC, Martin Steinegger, Sergey Ovchinnikov, Fajie Yuan
biorxiv; 28 May 2024
Abstract
Training and deploying large protein language models (PLMs) typically requires deep machine learning (ML) expertise—a significant barrier for many researchers outside the ML field. SaprotHub overcomes this challenge by offering an intuitive platform that democratizes every aspect of the PLM lifecycle—from training and prediction to storage and sharing. This approach fosters unprecedented collaboration within the biology community without demanding specialized ML skills. At SaprotHub’s core is Saprot, an advanced foundation PLM. Its ColabSaprot framework potentially powers hundreds of protein training and prediction applications, enabling researchers to collaboratively build and share customized models. This ecosystem not only enhances user engagement but catalyzes community-driven innovation in the biology community.
Links
Protein design using structure-based residue preferences
David Ding, Ada Y Shaw, Sam Sinai, Nathan Rollins, Noam Prywes, David F Savage, Michael T Laub, Debora S. Marks
Nature Communications; 22 Feb 2024
Abstract
Recent developments in protein design rely on large neural networks with up to 100s of millions of parameters, yet it is unclear which residue dependencies are critical for determining protein function. Here, we show that amino acid preferences at individual residues—without accounting for mutation interactions—explain much and sometimes virtually all of the combinatorial mutation effects across 8 datasets (R2 ~ 78-98%). Hence, few observations (~100 times the number of mutated residues) enable accurate prediction of held-out variant effects (Pearson r > 0.80). We hypothesized that the local structural contexts around a residue could be sufficient to predict mutation preferences, and develop an unsupervised approach termed CoVES (Combinatorial Variant Effects from Structure). Our results suggest that CoVES outperforms not just model-free methods but also similarly to complex models for creating functional and diverse protein variants. CoVES offers an effective alternative to complicated models for identifying functional protein mutations.
Links
Machine Learning for Functional Protein Design
Pascal Notin, Nathan Rollins, Yarin Gal, Chris Sander, Debora Marks
Nature Biotechnology; 15 Feb 2024
Recent breakthroughs in AI coupled with the rapid accumulation of protein sequence and structure data have radically transformed computational protein design. New methods promise to escape the constraints of natural and laboratory evolution, accelerating the generation of proteins for applications in biotechnology and medicine. To make sense of the exploding diversity of machine learning approaches, we introduce a unifying framework that classifies models on the basis of their use of three core data modalities: sequences, structures and functional labels. We discuss the new capabilities and outstanding challenges for the practical design of enzymes, antibodies, vaccines, nanomachines and more. We then highlight trends shaping the future of this field, from large-scale assays to more robust benchmarks, multimodal foundation models, enhanced sampling strategies and laboratory automation.
Links
GPR161 structure uncovers the redundant role of sterol-regulated ciliary cAMP signaling in the Hedgehog pathway
Nicholas Hoppe, Simone Harrison, Sun-Hee Hwang, Ziwei Chen, Masha Karelina, Ishan Deshpande, Carl-Mikael Suomivuori, Vivek R. Palicharla, Samuel P. Berry, Philipp Tschaikner, Dominik Regele, Douglas F. Covey, Eduard Stefan, Debora S. Marks, Jeremy F. Reiter, Ron O. Dror, Alex S. Evers, Saikat Mukhopadhyay & Aashish Manglik
Nature Structural & Molecular Biology; 7 Feb 2024
The orphan G protein-coupled receptor (GPCR) GPR161 plays a central role in development by suppressing Hedgehog signaling. The fundamental basis of how GPR161 is activated remains unclear. Here, we determined a cryogenic-electron microscopy structure of active human GPR161 bound to heterotrimeric Gs. This structure revealed an extracellular loop 2 that occupies the canonical GPCR orthosteric ligand pocket. Furthermore, a sterol that binds adjacent to transmembrane helices 6 and 7 stabilizes a GPR161 conformation required for Gs coupling. Mutations that prevent sterol binding to GPR161 suppress Gs-mediated signaling. These mutants retain the ability to suppress GLI2 transcription factor accumulation in primary cilia, a key function of ciliary GPR161. By contrast, a protein kinase A-binding site in the GPR161 C terminus is critical in suppressing GLI2 ciliary accumulation. Our work highlights how structural features of GPR161 interface with the Hedgehog pathway and sets a foundation to understand the role of GPR161 function in other signaling pathways.
Links
scPerturb: harmonized single-cell perturbation data
Stefan Peidli, Tessa D Green, Ciyue Shen, Torsten Gross, Joseph Min, Samuele Garda, Bo Yuan, Linus J Schumacher, Jake P Taylor-King, Debora S Marks, Augustin Luna, Nils Blüthgen, Chris Sander
Nature Methods; 26 January 2024
Abstract
Analysis across a growing number of single-cell perturbation datasets is hampered by poor data interoperability. To facilitate development and benchmarking of computational methods, we collect a set of 44 publicly available single-cell perturbation–response datasets with molecular readouts, including transcriptomics, proteomics and epigenomics. We apply uniform quality control pipelines and harmonize feature annotations. The resulting information resource, scPerturb, enables development and testing of computational methods, and facilitates comparison and integration across datasets. We describe energy statistics (E-statistics) for quantification of perturbation effects and significance testing, and demonstrate E-distance as a general distance measure between sets of single-cell expression profiles. We illustrate the application of E-statistics for quantifying similarity and efficacy of perturbations. The perturbation–response datasets and E-statistics computation software are publicly available at scperturb.org. This work provides an information resource for researchers working with single-cell perturbation data and recommendations for experimental design, including optimal cell counts and read depth.
Links
2023
ProteinNPT: Improving Protein Property Prediction and Design with Non-Parametric Transformers
Pascal Notin, Ruben Weitzman, Debora Marks, Yarin Gal
NeurIPS 2023; Dec 2023
Abstract
Protein design holds immense potential for optimizing naturally occurring proteins, with broad applications in drug discovery, material design, and sustainability. However, computational methods for protein engineering are confronted with significant challenges, such as an expansive design space, sparse functional regions, and a scarcity of available labels. These issues are further exacerbated in practice by the fact most real-life design scenarios necessitate the simultaneous optimization of multiple properties. In this work, we introduce ProteinNPT, a non-parametric transformer variant tailored to protein sequences and particularly suited to label-scarce and multi-task learning settings. We first focus on the supervised fitness prediction setting and develop several cross-validation schemes which support robust performance assessment. We subsequently reimplement prior top-performing baselines, introduce several extensions of these baselines by integrating diverse branches of the protein engineering literature, and demonstrate that ProteinNPT consistently outperforms all of them across a diverse set of protein property prediction tasks. Finally, we demonstrate the value of our approach for iterative protein design across extensive in silico Bayesian optimization and conditional sampling experiments.
Links
ProteinGym: Large-scale Benchmarks for Protein Fitness Prediction and Design
Pascal Notin, Aaron W Kollasch, Daniel Ritter, Lood van Niekerk, Steffanie Paul, Han Spinner, Nathan J Rollins, Ada Shaw, Rose Orenbuch, Ruben Weitzman, Jonathan Frazer, Mafalda Dias, Dinko Franceschi, Yarin Gal, Debora S Marks
NeurIPS 2023
Abstract
Predicting the effects of mutations in proteins is critical to many applications, from understanding genetic disease to designing novel proteins to address our most pressing challenges in climate, agriculture and healthcare. Despite an increase in machine learning-based protein modeling methods, assessing their effectiveness is problematic due to the use of distinct, often contrived, experimental datasets and variable performance across different protein families. Addressing these challenges requires scale. To that end we introduce ProteinGym v1.0, a large-scale and holistic set of benchmarks specifically designed for protein fitness prediction and design. It encompasses both a broad collection of over 250 standardized deep mutational scanning assays, spanning millions of mutated sequences, as well as curated clinical datasets providing high-quality expert annotations about mutation effects. We devise a robust evaluation framework that combines metrics for both fitness prediction and design, factors in known limitations of the underlying experimental methods, and covers both zero-shot and supervised settings. We report the performance of a diverse set of over 40 high-performing models from various subfields (eg., mutation effects, inverse folding) into a unified benchmark. We open source the corresponding codebase, datasets, MSAs, structures, predictions and develop a user-friendly website that facilitates comparisons across all settings.
Links
Combining Structure and Sequence for Superior Fitness Prediction
Steffanie Paul, Aaron Kollasch, Pascal Notin, Debora Marks
NeurIPS 2023 GenBio Workshop; Dec 2023
Abstract
Deep generative models of protein sequence and inverse folding models have shown great promise as protein design methods. While sequence-based models have shown strong zero-shot mutation effect prediction performance, inverse folding models have not been extensively characterized in this way. As these models use information from protein structures, it is likely that inverse folding models possess inductive biases that make them better predictors of certain function types. Using the collection of model scores contained in the newly updated ProteinGym, we systematically explore the differential zero-shot predictive power of sequence and inverse folding models. We find that inverse folding models consistently outperform the best-in-class sequence models on assays of protein thermostability, but have lower performance on other properties. Motivated by these findings, we develop StructSeq, an ensemble model combining information from sequence, multiple sequence alignments (MSAs), and structure. StructSeq achieves state-of-the-art Spearman correlation on ProteinGym and is robust to different functional assay types.
Links
An Energy Based Model for Incorporating Sequence Priors for Target-Specific Antibody Design
Yining Huang, Steffanie Paul, Debora Marks
NeurIPS 2023 GenBio Workshop; Dec 2023
Abstract
With the growing demand for antibody therapeutics, there is a great need for computational methods to accelerate antibody discovery and optimization. Advances in machine learning on graphs have been leveraged to develop generative models of antibody sequence and structure that condition on specific antigen epitopes. However, the data availability for training models on structure (∼5k antibody binding complexes Schneider et al. [2022]) is dwarfed by the amount of antibody sequence data available (> 550M sequences Olsen et al. [2022]) which have been used to train protein language models useful for antibody generation and optimization Here we motivate the combination of well-trained antibody sequence models and graph generative models on target structures to enhance their performance for target-conditioned antibody design. First, we present the results of an investigation into the sitewise design performance of popular target-conditioned design models. We show that target-conditioned models may not be incorporating target information into the generation of middle loop residues of the complementarity-determining region of the antibody sequence. Next, we propose an energy-based model framework designed to encourage a model to learn target-specific information by supplementing it with pre-trained marginal-sequence information. We present preliminary results on the development of this model and outline future steps to improve the model framework.
Links
Learning from pre-pandemic data to forecast viral antibody escape
Nicole N. Thadani, Sarah Gurev, Pascal Notin, Noor Youssef, Nathan J. Rollins, Daniel Ritter, Chris Sander, Yarin Gal, Debora S. Marks
Nature; 11 October 2023
Abstract
From early detection of variants of concern to vaccine and therapeutic design, pandemic preparedness depends on identifying viral mutations that escape the response of the host immune system. While experimental scans are useful for quantifying escape potential, they remain laborious and impractical for exploring the combinatorial space of mutations. Here we introduce a biologically grounded model to quantify the viral escape potential of mutations at scale. Our method - EVEscape - brings together fitness predictions from evolutionary models, structure-based features that assess antibody binding potential, and distances between mutated and wild-type residues. Unlike other models that predict variants of concern based on newly observed variants, EVEscape has no reliance on recent community prevalence, and is applicable before surveillance sequencing or experimental scans are broadly available. We validate EVEscape predictions against experimental data on H1N1, HIV and SARS-CoV-2, including data on immune escape. For SARS-CoV-2, we show that EVEscape anticipates mutation frequency, strain prevalence, and escape mutations. Drawing from GISAID, we provide continually updated escape predictions for all current strains of SARS-CoV-2.
Links
Removing bias in sequence models of protein fitness
Ada Shaw, Aviv Spinner, June Shin, Sarah Gurev, Nathan Rollins, Debora S Marks
biorxiv; 30 September 2023
Abstract
Unsupervised sequence models for protein fitness have emerged as powerful tools for protein design in order to engineer therapeutics and industrial enzymes, yet they are strongly biased towards potential designs that are close to their training data. This hinders their ability to generate functional sequences that are far away from natural sequences, as is often desired to design new functions. To address this problem, we introduce a de-biasing approach that enables the comparison of protein sequences across mutational depths to overcome the extant sequence similarity bias in natural sequence models. We demonstrate our method’s effectiveness at improving the relative natural sequence model predictions of experimentally measured variant functions across mutational depths. Using case studies proteins with very low functional percentages further away from the wild type, we demonstrate that our method improves the recovery of top-performing variants in these sparsely functional regimes. Our method is generally applicable to any unsupervised fitness prediction model, and for any function for any protein, and can thus easily be incorporated into any computational protein design pipeline. These studies have the potential to develop more efficient and cost-effective computational methods for designing diverse functional proteins and to inform underlying experimental library design to best take advantage of machine learning capabilities.
Links
A kernelized Stein discrepancy for biological sequences
Alan Nawzad Amin, Eli Nathan Weinstein, Debora S Marks
International Conference on Machine Learning; 23 July 2023
Abstract
Generative models of biological sequences are a powerful tool for learning from complex sequence data, predicting the effects of mutations, and designing novel biomolecules with desired properties. To evaluate generative models it is important to accurately measure differences between high-dimensional distributions. In this paper we propose the "KSD-B", a novel divergence measure for distributions over biological sequences that is based on the kernelized Stein discrepancy (KSD). The KSD-B can be evaluated even when the normalizing constant of the model is unknown; it allows for variable length sequences and can take into account biological notions of sequence distance. Unlike previous KSDs over discrete spaces the KSD-B (a) is theoretically guaranteed to detect convergence and non-convergence of distributions over sequence space and (b) can be efficiently estimated in practice. We demonstrate the advantages of the KSD-B on problems with synthetic and real data, and apply it to measure the fit of state-of-the-art machine learning models. Overall, the KSD-B enables rigorous evaluation of generative biological sequence models, allowing the accuracy of models, sampling procedures, and library designs to be checked reliably.
Links
An Atlas of Variant Effects to understand the genome at nucleotide resolution
Douglas M. Fowler, David J. Adams, Anna L. Gloyn, William C. Hahn, Debora S. Marks, Lara A. Muffley, James T. Neal, Frederick P. Roth, Alan F. Rubin, Lea M. Starita, Matthew E. Hurles
Genome Biology; 03 July 2023
Abstract
Sequencing has revealed hundreds of millions of human genetic variants, and continued efforts will only add to this variant avalanche. Insufficient information exists to interpret the effects of most variants, limiting opportunities for precision medicine and comprehension of genome function. A solution lies in experimental assessment of the functional effect of variants, which can reveal their biological and clinical impact. However, variant effect assays have generally been undertaken reactively for individual variants only after and, in most cases long after, their first observation. Now, multiplexed assays of variant effect can characterise massive numbers of variants simultaneously, yielding variant effect maps that reveal the function of every possible single nucleotide change in a gene or regulatory element. Generating maps for every protein encoding gene and regulatory element in the human genome would create an ‘Atlas’ of variant effect maps and transform our understanding of genetics and usher in a new era of nucleotide-resolution functional knowledge of the genome. An Atlas would reveal the fundamental biology of the human genome, inform human evolution, empower the development and use of therapeutics and maximize the utility of genomics for diagnosing and treating disease. The Atlas of Variant Effects Alliance is an international collaborative group comprising hundreds of researchers, technologists and clinicians dedicated to realising an Atlas of Variant Effects to help deliver on the promise of genomics.
Links
Simultaneous enhancement of multiple functional properties using evolution-informed protein design
Benjamin Fram, Ian Truebridge, Yang su, Adam J Riesselman, John B Ingraham, Alessandro Passera, Eve Napier, Nicole N Thadani, Samuel Lim, Kristen Roberts, Gurleen Kaur, Michael Stiffler, Debora S Marks, Christopher D Bahl, Amir R Khan, Chris Sander, Nicholas P Gauthier
PubMed Preprint; 09 May 2023
Abstract
Designing optimized proteins is important for a range of practical applications. Protein design is a rapidly developing field that would benefit from approaches that enable many changes in the amino acid primary sequence, rather than a small number of mutations, while maintaining structure and enhancing function. Homologous protein sequences contain extensive information about various protein properties and activities that have emerged over billions of years of evolution. Evolutionary models of sequence co-variation, derived from a set of homologous sequences, have proven effective in a range of applications including structure determination and mutation effect prediction. In this work we apply one of these models (EVcouplings) to computationally design highly divergent variants of the model protein TEM-1 β-lactamase, and characterize these designs experimentally using multiple biochemical and biophysical assays. Nearly all designed variants were functional, including one with 84 mutations from the nearest natural homolog. Surprisingly, all functional designs had large increases in thermostability and most had a broadening of available substrates. These property enhancements occurred while maintaining a nearly identical structure to the wild type enzyme. Collectively, this work demonstrates that evolutionary models of sequence co-variation (1) are able to capture complex epistatic interactions that successfully guide large sequence departures from natural contexts, and (2) can be applied to generate functional diversity useful for many applications in protein design.
Links
A deep learning algorithm to predict risk of pancreatic cancer from disease trajectories
Davide Placido, Bo Yuan, Jessica X. Hjatelin, Chunlei Zheng, Amalie D. Haue, Piotr J. Chmura, Chen Yuan, Jihye Kim, Renato Umeton, Gregory Antell, Alexander Chowdhury, Alexandra Franz, Lauren Brais, Elizabeth Andrew, Debora S. Marks, Aviv Regev, Siamack Ayandeh, Mary T. Brophy, Nhan V. Do, Peter Kraft, Brian M. Wolpin, Michael H. Rosenthal, Nathanael R. Fillmore, Soren Brunak, Chris Sander
Nature Medicine; 08 May 2023
Abstract
Pancreatic cancer is an aggressive disease that typically presents late with poor outcomes, indicating a pronounced need for early detection. In this study, we applied artificial intelligence methods to clinical data from 6 million patients (24,000 pancreatic cancer cases) in Denmark (Danish National Patient Registry (DNPR)) and from 3 million patients (3,900 cases) in the United States (US Veterans Affairs (US-VA)). We trained machine learning models on the sequence of disease codes in clinical histories and tested prediction of cancer occurrence within incremental time windows (CancerRiskNet). For cancer occurrence within 36 months, the performance of the best DNPR model has area under the receiver operating characteristic (AUROC) curve = 0.88 and decreases to AUROC (3m) = 0.83 when disease events within 3 months before cancer diagnosis are excluded from training, with an estimated relative risk of 59 for 1,000 highest-risk patients older than age 50 years. Cross-application of the Danish model to US-VA data had lower performance (AUROC = 0.71), and retraining was needed to improve performance (AUROC = 0.78, AUROC (3m) = 0.76). These results improve the ability to design realistic surveillance programs for patients at elevated risk, potentially benefiting lifespan and quality of life by early detection of this aggressive cancer.
Links
The relaxin receptor RXFP1 signals through a mechanism of autoinhibition
Sarah C. Erlandson, Shaun Rawson, James Osei-Owusu, Kelly P. Brock, Xinyue Liu, Joao A. Paulo, Julian Mintseris, Steven P. Gygi, Debora S. Marks, Xiaojing Cong, Andrew C. Kruse
Nature Chemical Biology; 20 April 2023
Abstract
The relaxin family peptide receptor 1 (RXFP1) is the receptor for relaxin-2, an important regulator of reproductive and cardiovascular physiology. RXFP1 is a multi-domain G protein-coupled receptor (GPCR) with an ectodomain consisting of a low-density lipoprotein receptor class A (LDLa) module and leucine-rich repeats. The mechanism of RXFP1 signal transduction is clearly distinct from that of other GPCRs, but remains very poorly understood. In the present study, we determine the cryo-electron microscopy structure of active-state human RXFP1, bound to a single-chain version of the endogenous agonist relaxin-2 and the heterotrimeric Gs protein. Evolutionary coupling analysis and structure-guided functional experiments reveal that RXFP1 signals through a mechanism of autoinhibition. Our results explain how an unusual GPCR family functions, providing a path to rational drug development targeting the relaxin receptors.
Links
Coordination of bacterial cell wall and outer membrane biosynthesis
Katherine R Hummels, Samuel P Berry, Zhaoqi Li, Atsushi Taguchi, Joseph K Min, Suzanne Walker, Debora S Marks, Thomas G Bernhardt
Nature; 01 March 2023
Abstract
Gram-negative bacteria surround their cytoplasmic membrane with a peptidoglycan (PG) cell wall and an outer membrane (OM) with an outer leaflet composed of lipopolysaccharide (LPS)1. This complex envelope presents a formidable barrier to drug entry and is a major determinant of the intrinsic antibiotic resistance of these organisms2. The biogenesis pathways that build the surface are also targets of many of our most effective antibacterial therapies3. Understanding the molecular mechanisms underlying the assembly of the Gram-negative envelope therefore promises to aid the development of new treatments effective against the growing problem of drug-resistant infections. Although the individual pathways for PG and OM synthesis and assembly are well characterized, almost nothing is known about how the biogenesis of these essential surface layers is coordinated. Here we report the discovery of a regulatory interaction between the committed enzymes for the PG and LPS synthesis pathways in the Gram-negative pathogen Pseudomonas aeruginosa. We show that the PG synthesis enzyme MurA interacts directly and specifically with the LPS synthesis enzyme LpxC. Moreover, MurA was shown to stimulate LpxC activity in cells and in a purified system. Our results support a model in which the assembly of the PG and OM layers in many proteobacterial species is coordinated by linking the activities of the committed enzymes in their respective synthesis pathways.
Links
2022
An In Silico Method to Assess Antibody Fragment Polyreactivity
Edward P Harvey, Jung-Eun Shin, Meredith A Skiba, Genevieve R Nemeth, Joseph D Hurley, Alon Wellner, Ada Y Shaw, Victor G Miranda, Joseph K Min, Chang C Liu, Debora S Marks, Andrew C Kruse
Nature Communications; 07 Dec 2022
Abstract
Antibodies are essential biological research tools and important therapeutic agents, but some exhibit non-specific binding to off-target proteins and other biomolecules. Such polyreactive antibodies compromise screening pipelines, lead to incorrect and irreproducible experimental results, and are generally intractable for clinical development. Here, we design a set of experiments using a diverse naïve synthetic camelid antibody fragment (nanobody) library to enable machine learning models to accurately assess polyreactivity from protein sequence (AUC > 0.8). Moreover, our models provide quantitative scoring metrics that predict the effect of amino acid substitutions on polyreactivity. We experimentally test our models’ performance on three independent nanobody scaffolds, where over 90% of predicted substitutions successfully reduced polyreactivity. Importantly, the models allow us to diminish the polyreactivity of an angiotensin II type I receptor antagonist nanobody, without compromising its functional properties. We provide a companion web-server that offers a straightforward means of predicting polyreactivity and polyreactivity-reducing mutations for any given nanobody sequence.
Links
Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval
Pascal Notin, Mafalda Dias, Jonathan Frazer, Javier Marchena-Hurtado, Aidan Gomez, Debora S. Marks, Yarin Gal
arXiv; 27 May 2022
Abstract
The ability to accurately model the fitness landscape of protein sequences is critical to a wide range of applications, from quantifying the effects of human variants on disease likelihood, to predicting immune-escape mutations in viruses and designing novel biotherapeutic proteins. Deep generative models of protein sequences trained on multiple sequence alignments have been the most successful approaches so far to address these tasks. The performance of these methods is however contingent on the availability of sufficiently deep and diverse alignments for reliable training. Their potential scope is thus limited by the fact many protein families are hard, if not impossible, to align. Large language models trained on massive quantities of non-aligned protein sequences from diverse families address these problems and show potential to eventually bridge the performance gap. We introduce Tranception, a novel transformer architecture leveraging autoregressive predictions and retrieval of homologous sequences at inference to achieve state-of-the-art fitness prediction performance. Given its markedly higher performance on multiple mutants, robustness to shallow alignments and ability to score indels, our approach offers significant gain of scope over existing approaches. To enable more rigorous model testing across a broader range of protein families, we develop ProteinGym -- an extensive set of multiplexed assays of variant effects, substantially increasing both the number and diversity of assays compared to existing benchmarks.
Links
Natural and designed proteins inspired by extremotolerant organisms can form condensates and attenuate apoptosis in human cells
Mike T Veling, Dan T Nguyen, Nicole N Thadani, Michela E Oster, Nathan J Rollins, Kelly P Brock, Neville P Bethel, Samuel Lim, David Baker, Jeffrey C Way, Debora S Marks, Roger L Chang, Pamela A Silver
ACS Synthethic Biology; 18 Feb 2022
Abstract
Many organisms can survive extreme conditions and successfully recover to normal life. This extremotolerant behavior has been attributed in part to repetitive, amphipathic, and intrinsically disordered proteins that are upregulated in the protected state. Here, we assemble a library of approximately 300 naturally-occurring and designed extremotolerance-associated proteins to assess their ability to protect human cells from chemically-induced apoptosis. We show that proteins from tardigrades, nematodes, and the Chinese giant salamander are apoptosis protective. Notably, we identify a region of the human ApoE protein with similarity to extremotolerance-associated proteins that also protects against apoptosis. This region mirrors the phase separation behavior seen with such proteins, like the tardigrade protein CAHS2. Moreover, we identify a synthetic protein, DHR81, that shares this combination of elevated phase separation propensity and apoptosis protection. Finally, we demonstrate that driving protective proteins into the condensate state increases apoptosis protection, and highlight the ability for DHR81 condensates to sequester caspase-7. Taken together, this work draws a link between extremotolerance-associated proteins, condensate formation, and human cellular protection.
Links
Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval
Pascal Notin, Mafalda Dias, Jonathan Frazer, Javier Marchena-Hurtado, Aidan Gomez, Debora S. Marks, Yarin Gal
arXiv; 27 May 2022
Abstract
The ability to accurately model the fitness landscape of protein sequences is critical to a wide range of applications, from quantifying the effects of human variants on disease likelihood, to predicting immune-escape mutations in viruses and designing novel biotherapeutic proteins. Deep generative models of protein sequences trained on multiple sequence alignments have been the most successful approaches so far to address these tasks. The performance of these methods is however contingent on the availability of sufficiently deep and diverse alignments for reliable training. Their potential scope is thus limited by the fact many protein families are hard, if not impossible, to align. Large language models trained on massive quantities of non-aligned protein sequences from diverse families address these problems and show potential to eventually bridge the performance gap. We introduce Tranception, a novel transformer architecture leveraging autoregressive predictions and retrieval of homologous sequences at inference to achieve state-of-the-art fitness prediction performance. Given its markedly higher performance on multiple mutants, robustness to shallow alignments and ability to score indels, our approach offers significant gain of scope over existing approaches. To enable more rigorous model testing across a broader range of protein families, we develop ProteinGym -- an extensive set of multiplexed assays of variant effects, substantially increasing both the number and diversity of assays compared to existing benchmarks.
Links
2021
A Structured Observation Distribution for Generative Biological Sequence Prediction and Forecasting
Eli N Weinstein, Debora S Marks
Proceedings of the 38th International Conference on Machine Learning; 2021
Abstract
Generative probabilistic modeling of biological sequences has widespread existing and potential application across biology and biomedicine, from evolutionary biology to epidemiology to protein design. Many standard sequence analysis methods preprocess data using a multiple sequence alignment (MSA) algorithm, one of the most widely used computational methods in all of science. However, as we show in this article, training generative probabilistic models with MSA preprocessing leads to statistical pathologies in the context of sequence prediction and forecasting. To address these problems, we propose a principled drop-in alternative to MSA preprocessing in the form of a structured observation distribution (the "MuE" distribution). We prove theoretically that the MuE distribution comprehensively generalizes popular methods for inferring biological sequence alignments, and provide a precise characterization of how such biological models have differed from natural language latent alignment models. We show empirically that models that use the MuE as an observation distribution outperform comparable methods across a variety of datasets, and apply MuE models to a novel problem for generative probabilistic sequence models: forecasting pathogen evolution.
Links
MaveDB v2: a curated community database with over three million variant effects from multiplexed functional assays
Alan F Rubin, Joseph K Min, Nathan J Rollins, Estelle Y Da, Daniel Esposito, Matthew Harrington, Jeremy Stone, Aisha Haley Bianchi, Mafalda Dias, Jonathan Frazer, Yunfan Fu, Molly Gallaher, Iris Li, Olivia Moscatelli, Jesslyn YL Ong, Joshua E Rollins, Matthew J Wakefield, Shenyi “Sunny” Ye, Amy Tam, Abbye E McEwen, Lea M Starita, Vanessa L Bryant, Debora S Marks, Douglas M Fowler
bioRxiv; 30 November 2021
Abstract
A central problem in genomics is understanding the effect of individual DNA variants. Multiplexed Assays of Variant Effect (MAVEs) can help address this challenge by measuring all possible single nucleotide variant effects in a gene or regulatory sequence simultaneously. Here we describe MaveDB v2, which has become the database of record for MAVEs. MaveDB now contains a large fraction of published studies, comprising over two hundred datasets and three million variant effect measurements. We created tools and APIs to streamline data submission and access, transforming MaveDB into a hub for the analysis and dissemination of these impactful datasets.
Links
Optimal Design of Stochastic DNA Synthesis Protocols based on Generative Sequence Models
Eli N Weinstein, Alan N Amin, Will Grathwohl, Daniel Kassler, Jean Disset, Debora S Marks
bioRxiv; 28 Oct 2021
Abstract
Generative probabilistic models of biological sequences have widespread existing and potential applications in analyzing, predicting and designing proteins, RNA and genomes. To test the predictions of such a model experimentally, the standard approach is to draw samples, and then synthesize each sample individually in the laboratory. However, often orders of magnitude more sequences can be experimentally assayed than can affordably be synthesized individually. In this article, we propose instead to use stochastic synthesis methods, such as mixed nucleotides or trimers. We describe a black-box algorithm for optimizing stochastic synthesis protocols to produce approximate samples from any target generative model. We establish theoretical bounds on the method’s performance, and validate it in simulation using held-out sequence-to-function predictors trained on real experimental data. We show that using optimized stochastic synthesis protocols in place of individual synthesis can increase the number of hits in protein engineering efforts by orders of magnitude, e.g. from zero to a thousand.
Links
Dormant spores sense amino acids through the B subunits of their germination receptors
Lior Artzi, Assaf Alon, Kelly P Brock, Anna G Green, Amy Tam, Fernando H Ramírez-Guadiana, Debora S Marks, Andrew Kruse, David Z Rudner
bioRxiv; 28 Oct 2021
Abstract
Bacteria from the orders Bacillales and Clostridiales differentiate into stress-resistant spores that can remain dormant for years, yet rapidly germinate upon nutrient sensing. How spores monitor nutrients is poorly understood but in most cases requires putative membrane receptors. The prototypical receptor from Bacillus subtilis consists of three proteins (GerAA, GerAB, GerAC) required for germination in response to L-alanine. GerAB belongs to the Amino Acid-Polyamine-Organocation superfamily of transporters. Using evolutionary co-variation analysis, we provide evidence that GerAB adopts a structure similar to an L-alanine transporter from this superfamily. We show that mutations in gerAB predicted to disrupt the ligand-binding pocket impair germination, while mutations predicted to function in L-alanine recognition enable spores to respond to L-leucine or L-serine. Finally, substitutions of bulkier residues at these positions cause constitutive germination. These data suggest that GerAB is the L-alanine sensor and that B subunits in this broadly conserved family function in nutrient detection.
Links
Disease variant prediction with deep generative models of evolutionary data
Jonathan Frazer, Pascal Notin, Mafalda Dias, Aidan Gomez, Joseph K Min, Kelly Brock, Yarin Gal, Debora S Marks
Nature;
27 Oct 2021
Abstract
Quantifying the pathogenicity of protein variants in human disease-related genes would have a marked effect on clinical decisions, yet the overwhelming majority (over 98%) of these variants still have unknown consequences. In principle, computational methods could support the large-scale interpretation of genetic variants. However, state-of-the-art methods have relied on training machine learning models on known disease labels. As these labels are sparse, biased and of variable quality, the resulting models have been considered insufficiently reliable. Here we propose an approach that leverages deep generative models to predict variant pathogenicity without relying on labels. By modelling the distribution of sequence variation across organisms, we implicitly capture constraints on the protein sequences that maintain fitness. Our model EVE (evolutionary model of variant effect) not only outperforms computational approaches that rely on labelled data but also performs on par with, if not better than, predictions from high-throughput experiments, which are increasingly used as evidence for variant classification. We predict the pathogenicity of more than 36 million variants across 3,219 disease genes and provide evidence for the classification of more than 256,000 variants of unknown significance. Our work suggests that models of evolutionary information can provide valuable independent evidence for variant interpretation that will be widely useful in research and clinical settings.
Links
Coevolution of interacting proteins through non-contacting and non-specific mutations
David Ding, Anna G Green, Boyuan Wang, Thuy-Lan Vo Lite, Eli N Weinstein, Debora S Marks, Michael T Laub
bioRxiv;
08 Oct 2021
Abstract
Proteins often accumulate neutral mutations that do not affect current functions but can profoundly influence future mutational possibilities and functions. Understanding such hidden potential has major implications for protein design and evolutionary forecasting, but has been limited by a lack of systematic efforts to identify potentiating mutations. Here, through the comprehensive analysis of a bacterial toxin-antitoxin system, we identified all possible single substitutions in the toxin that enable it to tolerate otherwise interface-disrupting mutations in its antitoxin. Strikingly, the majority of enabling mutations in the toxin do not contact, and promote tolerance non-specifically to, many different antitoxin mutations, despite covariation in homologs occurring primarily between specific pairs of contacting residues across the interface. In addition, the enabling mutations we identified expand future mutational paths that both maintain old toxin-antitoxin interactions and form new ones. These non-specific mutations are missed by widely used covariation and machine learning methods. Identifying such enabling mutations will be critical for ensuring continued binding of therapeutically relevant proteins, such as antibodies, aimed at evolving targets.
Links
Natural and designed proteins inspired by extremotolerant organisms can form condensates and attenuate apoptosis in human cells
Mike T. Veling, Dan T. Nguyen, Nicole N. Thadani, Michela E. Oster, Nathan J. Rollins, Kelly P. Brock, Neville P. Bethel, David Baker, Jeffrey C. Way, Debora S. Marks, Roger L. Chang, Pamela A. Silver
bioRxiv;
01 Oct 2021
Abstract
Many organisms can survive extreme conditions and successfully recover to normal life. This extremotolerant behavior has been attributed in part to repetitive, amphipathic, and intrinsically disordered proteins that are upregulated in the protected state. Here, we assemble a library of approximately 300 naturally-occurring and designed extremotolerance-associated proteins to assess their ability to protect human cells from chemically-induced apoptosis. We show that proteins from tardigrades, nematodes, and the Chinese giant salamander are apoptosis protective. Notably, we identify a region of the human ApoE protein with similarity to extremotolerance-associated proteins that also protects against apoptosis. This region mirrors the phase separation behavior seen with such proteins, like the tardigrade protein CAHS2. Moreover, we identify a synthetic protein, DHR81, that shares this combination of elevated phase separation propensity and apoptosis protection. Finally, we demonstrate that driving protective proteins into the condensate state increases apoptosis protection, and highlight the ability for DHR81 condensates to sequester caspase-7. Taken together, this work draws a link between extremotolerance-associated proteins, condensate formation, and human cellular protection.
Links
A generative nonparametric Bayesian model for whole genomes
Alan N Amin*, Eli N Weinstein*, Debora S Marks
*These authors contributed equally.
NeurIPS 2021;
28 Sep 2021
Abstract
Generative probabilistic modeling of biological sequences has widespread existing and potential use across biology and biomedicine, particularly given advances in high-throughput sequencing, synthesis and editing. However, we still lack methods with nucleotide resolution that are tractable at the scale of whole genomes and that can achieve high predictive accuracy either in theory or practice. In this article we propose a new generative sequence model, the Bayesian embedded autoregressive (BEAR) model, which uses a parametric autoregressive model to specify a conjugate prior over a nonparametric Bayesian Markov model. We explore, theoretically and empirically, applications of BEAR models to a variety of statistical problems including density estimation, robust parameter estimation, goodness-of-fit tests, and two-sample tests. We prove rigorous asymptotic consistency results including nonparametric posterior concentration rates. We scale inference in BEAR models to datasets containing tens of billions of nucleotides. On genomic, transcriptomic, and metagenomic sequence data we show that BEAR models provide large increases in predictive performance as compared to parametric autoregressive models, among other results. BEAR models offer a flexible and scalable framework, with theoretical guarantees, for building and critiquing generative models at the whole genome scale.
Links
Pancreatic cancer risk predicted from disease trajectories using deep learning
Davide Placido, Bo Yuan, Jessica X Hjaltelin, Amalie D Haue, Piotr J Chmura, Chen Yuan, Jihye Kim, Renato Umeton, Gregory Antell, Alexander Chowdhury, Alexandra Franz, Lauren Brais, Elizabeth Andrews, Debora S Marks, Aviv Regev, Peter Kraft, Brian M Wolpin, Michael Rosenthal, Søren Brunak, Chris Sander
BioRxiv; 28 June 2021
Abstract
Pancreatic cancer is an aggressive disease that typically presents late with poor patient outcomes. There is a pronounced medical need for early detection of pancreatic cancer, which can be facilitated by identifying high-risk populations. Here we apply artificial intelligence (AI) methods to a large corpus of more than 6 million patient records spanning 40 years with 24,000 pancreatic cancer cases in the Danish National Patient Registry. In contrast to existing methods that do not use temporal information, we explicitly train machine learning models on the time sequence of diseases in patient clinical histories. In addition, the models predict the risk of cancer occurrence in time intervals of 3 to 60 months duration after risk assessment. For cancer occurrence within 12 months, the performance of the best model trained on full trajectories (AUROC=0.91) substantially exceeds that of a model without time information (AUROC=0.81). For the best model, lower performance (AUROC=0.86) results when disease events within a 3 month window before cancer diagnosis are excluded from training, reflecting the decreasing information value of earlier disease events. These results raise the state-of-the-art level of performance of cancer risk prediction on real-world data sets and provide support for the design of real-world population-wide clinical screening trials, in which high risk patients are assigned to serial imaging and measurement of blood-based markers to facilitate earlier cancer detection. AI on real-world clinical records has the potential to shift focus from treatment of late- to early-stage cancer, benefiting patients by improving lifespan and quality of life.
Links
Rapid generation of potent antibodies by autonomous hypermutation in yeast
Alon Wellner, Conor McMahon, Morgan SA Gilman, Jonathan R Clements, Sarah Clark, Kianna M Nguyen, Ming H Ho, Vincent J Hu, Jung-Eun Shin, Jared Feldman, Blake M Hauser, Timothy M Caradonna, Laura M Wingler, Aaron G Schmidt, Debora S Marks, Jonathan Abraham, Andrew C Kruse, Chang C Liu
Nature chemical biology
; 24 Jun 2021
Abstract
The predominant approach for antibody generation remains animal immunization, which can yield exceptionally selective and potent antibody clones owing to the powerful evolutionary process of somatic hypermutation. However, animal immunization is inherently slow, not always accessible and poorly compatible with many antigens. Here, we describe ‘autonomous hypermutation yeast surface display’ (AHEAD), a synthetic recombinant antibody generation technology that imitates somatic hypermutation inside engineered yeast. By encoding antibody fragments on an error-prone orthogonal DNA replication system, surface-displayed antibody repertoires continuously mutate through simple cycles of yeast culturing and enrichment for antigen binding to produce high-affinity clones in as little as two weeks. We applied AHEAD to generate potent nanobodies against the SARS-CoV-2 S glycoprotein, a G-protein-coupled receptor and other targets, offering a template for streamlined antibody generation at large.
Links
Protein design and variant prediction using autoregressive generative models
Jung-Eun Shin*, Adam J Riesselman*, Aaron W Kollasch*, Conor McMahon, Elana Simon, Chris Sander, Aashish Manglik, Andrew C Kruse, Debora S Marks
*These authors contributed equally.
Nature communications;
23 Apr 2021
Abstract
The ability to design functional sequences and predict effects of variation is central to protein engineering and biotherapeutics. State-of-art computational methods rely on models that leverage evolutionary information but are inadequate for important applications where multiple sequence alignments are not robust. Such applications include the prediction of variant effects of indels, disordered proteins, and the design of proteins such as antibodies due to the highly variable complementarity determining regions. We introduce a deep generative model adapted from natural language processing for prediction and design of diverse functional sequences without the need for alignments. The model performs state-of-art prediction of missense and indel effects and we successfully design and test a diverse 105-nanobody library that shows better expression than a 1000-fold larger synthetic library. Our results demonstrate the power of the alignment-free autoregressive model in generalizing to regions of sequence space traditionally considered beyond the reach of prediction and design.
Links
Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences
Anna G Green*, Hadeer Elhabashy*, Kelly P Brock, Rohan Maddamsetti, Oliver Kohlbacher, Debora S Marks
*These authors contributed equally.
Nature communications;
02 Mar 2021
Abstract
Increasing numbers of protein interactions have been identified in high-throughput experiments, but only a small proportion have solved structures. Recently, sequence coevolution-based approaches have led to a breakthrough in predicting monomer protein structures and protein interaction interfaces. Here, we address the challenges of large-scale interaction prediction at residue resolution with a fast alignment concatenation method and a probabilistic score for the interaction of residues. Importantly, this method (EVcomplex2) is able to assess the likelihood of a protein interaction, as we show here applied to large-scale experimental datasets where the pairwise interactions are unknown. We predict 504 interactions de novo in the E. coli membrane proteome, including 243 that are newly discovered. While EVcomplex2 does not require available structures, coevolving residue pairs can be used to produce structural models of protein interactions, as done here for membrane complexes including the Flagellar Hook-Filament Junction and the Tol/Pal complex.
Links
CellBox: interpretable machine learning for perturbation biology with application to the design of cancer combination therapy
Bo Yuan, Ciyue Shen, Augustin Luna, Anil Korkut, Debora S Marks, John Ingraham, Chris Sander
Cell systems; 17 Feb 2021
Abstract
Systematic perturbation of cells followed by comprehensive measurements of molecular and phenotypic responses provides informative data resources for constructing computational models of cell biology. Models that generalize well beyond training data can be used to identify combinatorial perturbations of potential therapeutic interest. Major challenges for machine learning on large biological datasets are to find global optima in a complex multidimensional space and mechanistically interpret the solutions. To address these challenges, we introduce a hybrid approach that combines explicit mathematical models of cell dynamics with a machine-learning framework, implemented in TensorFlow. We tested the modeling framework on a perturbation-response dataset of a melanoma cell line after drug treatments. The models can be efficiently trained to describe cellular behavior accurately. Even though completely data driven and independent of prior knowledge, the resulting de novo network models recapitulate some known interactions. The approach is readily applicable to various kinetic models of cell biology.
Links
High-content screening of coronavirus genes for innate immune suppression reveals enhanced potency of SARS-CoV-2 proteins
Erika J Olson, David M Brown, Timothy Z Chang, Lin Ding, Tai L Ng, H Sloane Weiss, Peter Koch, Yukiye Koide, Nathan Rollins, Pia Mach, Tobias Meisinger, Trenton Bricken, Joshus Rollins, Yun Zhang, Colin Molloy, Briodget N Queenan, Timothy Mitchison, Debora Marks, Jeffrey C Way, John I Glass, Pamela A Silver
bioRxiv; 01 Jan 2021
Abstract
Suppression of the host intracellular innate immune system is an essential aspect of viral replication. Here, we developed a suite of medium-throughput high-content cell-based assays to reveal the effect of individual coronavirus proteins on antiviral innate immune pathways. Using these assays, we screened the 196 protein products of seven coronaviruses (SARS-CoV-2,SARS-CoV-1, 229E, NL63, OC43, HKU1 and MERS). This includes a previously unidentified gene in SARS-CoV-2 encoded within the Spike gene. We observe immune-suppressing activity in both known host-suppressing genes (e.g., NSP1, Orf6, NSP3, and NSP5) as well as other coronavirus genes, including the newly identified SARS-CoV-2 protein. Moreover, the genes encoded by SARS-CoV-2 are generally more potent immune suppressors than their homologues from the other coronaviruses. This suite of pathway-based and mechanism-agnostic assays could serve as the basis for rapid in vitro prediction of the pathogenicity of novel viruses based on provision of sequence information alone.
Links
A structured observation distribution for generative biological sequence prediction and forecasting
Eli N Weinstein, Debora S Marks
bioRxiv; 01 Jan 2021
Abstract
Generative probabilistic modeling of biological sequences has widespread existing and potential application across biology and biomedicine, from evolutionary biology to epidemiology to protein design. Many standard sequence analysis methods preprocess data using a multiple sequence alignment (MSA) algorithm, one of the most widely used computational methods in all of science. However, as we show in this article, training generative probabilistic models with MSA preprocessing leads to statistical pathologies in the context of sequence prediction and forecasting. To address these problems, we propose a principled drop-in alternative to MSA preprocessing in the form of a structured observation distribution (the "MuE" distribution). The MuE is a latent alignment model in which not only the alignment variable but also the regressor sequence can be latent. We prove theoretically that the MuE distribution comprehensively generalizes popular methods for inferring biological sequence alignments, and provide a precise characterization of how such biological models have differed from natural language latent alignment models. We show empirically that models that use the MuE as an observation distribution outperform comparable methods across a variety of datasets, and apply MuE models to a novel problem for generative probabilistic sequence models: forecasting pathogen evolution.
Links
2020
Large-scale clinical interpretation of genetic variants using evolutionary data and deep learning
Jonathan Frazer, Pascal Notin, Mafalda Dias, Aidan Gomez, Kelly Brock, Yarin Gal, Debora Marks
bioRxiv; 22 Dec 2020
Abstract
Quantifying the pathogenicity of protein variants in human disease-related genes would have a profound impact on clinical decisions, yet the overwhelming majority (over 98%) of these variants still have unknown consequences. In principle, computational methods could support the large-scale interpretation of genetic variants. However, prior methods have relied on training machine learning models on available clinical labels. Since these labels are sparse, biased, and of variable quality, the resulting models have been considered insufficiently reliable. By contrast, our approach leverages deep generative models to predict the clinical significance of protein variants without relying on labels. The natural distribution of protein sequences we observe across organisms is the result of billions of evolutionary experiments. By modeling that distribution, we implicitly capture constraints on the protein sequences that maintain fitness. Our model EVE (Evolutionary model of Variant Effect) not only outperforms computational approaches that rely on labelled data, but also performs on par, if not better than, high-throughput assays which are increasingly used as strong evidence for variant classification. After thorough validation on clinical labels, we predict the pathogenicity of 11 million variants across 1,081 disease genes, and assign high-confidence reclassification for 72k Variants of Unknown Significance. Our work suggests that models of evolutionary information can provide a strong source of independent evidence for variant interpretation and that the approach will be widely useful in research and clinical settings.
Links
Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact
Melissa A Chiasson, Nathan J Rollins, Jason J Stephany, Katherine A Sitko, Kenneth A Matreyek, Marta Verby, Song Sun, Frederick P Roth, Daniel DeSloover, Debora S Marks, Allan E Rettie, Douglas M Fowler
elife; 01 Sep 2020
Abstract
Vitamin K epoxide reductase (VKOR) drives the vitamin K cycle, activating vitamin K-dependent blood clotting factors. VKOR is also the target of the widely used anticoagulant drug, warfarin. Despite VKOR’s pivotal role in coagulation, its structure and active site remain poorly understood. In addition, VKOR variants can cause vitamin K-dependent clotting factor deficiency or alter warfarin response. Here, we used multiplexed, sequencing-based assays to measure the effects of 2,695 VKOR missense variants on abundance and 697 variants on activity in cultured human cells. The large-scale functional data, along with an evolutionary coupling analysis, supports a four transmembrane domain topology, with variants in transmembrane domains exhibiting strongly deleterious effects on abundance and activity. Functionally constrained regions of the protein define the active site, and we find that, of four conserved cysteines putatively critical for function, only three are absolutely required. Finally, 25% of human VKOR missense variants show reduced abundance or activity, possibly conferring warfarin sensitivity or causing disease.
Links
Interpretable machine learning for perturbation biology
Judy Shen, Bo Yuan, Augustin Luna, Anil Korkut, Debora Marks, John Ingraham, Chris Sander
Cancer Research; Published 15 Aug 2020
Abstract
Systematic perturbation of cells followed by comprehensive measurements of molecular and phenotypic responses provides an informative data resource for constructing computational models of cell biology. Models that generalize well beyond training data can be used to identify combinatorial perturbations of potential therapeutic interest. Major challenges for machine learning on large biological datasets are to find global optima in an enormously complex multi-dimensional solution space and to mechanistically interpret the solutions. To address these challenges, we introduce a hybrid approach that combines explicit mathematical models of dynamic cell biological processes with a machine learning framework, implemented in Tensorflow. We tested the modeling framework on a perturbation-response dataset for a melanoma cell line after drug treatments. The models can be efficiently trained to accurately describe cellular behavior, as tested by cross-validation. Even though completely data-driven and independent of prior knowledge, the resulting de novo network models recapitulate known interactions. The main predictive application of our work is the identification of combinatorial candidates for cancer therapy. This approach is readily applicable to a wide range of kinetic models of cell biology.
Links
Megan Sjodt, Patricia DA Rohs, Morgan SA Gilman, Sarah C Erlandson, Sanduo Zheng, Anna G Green, Kelly P Brock, Atsushi Taguchi, Daniel Kahne, Suzanne Walker, Debora S Marks, David Z Rudner, Thomas G Bernhardt, Andrew C Kruse
Nature microbiology; Published 09 Mar 2020
Structural coordination of polymerization and crosslinking by a SEDS–bPBP peptidoglycan synthase complex
Abstract
The shape, elongation, division and sporulation (SEDS) proteins are a highly conserved family of transmembrane glycosyltransferases that work in concert with class B penicillin-binding proteins (bPBPs) to build the bacterial peptidoglycan cell wall1,2,3,4,5,6. How these proteins coordinate polymerization of new glycan strands with their crosslinking to the existing peptidoglycan meshwork is unclear. Here, we report the crystal structure of the prototypical SEDS protein RodA from Thermus thermophilus in complex with its cognate bPBP at 3.3 Å resolution. The structure reveals a 1:1 stoichiometric complex with two extensive interaction interfaces between the proteins: one in the membrane plane and the other at the extracytoplasmic surface. When in complex with a bPBP, RodA shows an approximately 10 Å shift of transmembrane helix 7 that exposes a large membrane-accessible cavity. Negative-stain electron microscopy reveals that the complex can adopt a variety of different conformations. These data define the bPBP pedestal domain as the key allosteric activator of RodA both in vitro and in vivo, explaining how a SEDS–bPBP complex can coordinate its dual enzymatic activities of peptidoglycan polymerization and crosslinking to build the cell wall.
Links
Michael A Stiffler, Frank J Poelwijk, Kelly P Brock, Richard R Stein, Adam Riesselman, Joan Teyra, Sachdev S Sidhu, Debora S Marks, Nicholas P Gauthier, Chris Sander
Cell systems; Published 22 Jan 2020
Protein structure from experimental evolution
Abstract
Natural evolution encodes rich information about the structure and function of biomolecules in the genetic record. Previously, statistical analysis of co-variation patterns in natural protein families has enabled the accurate computation of 3D structures. Here, we explored generating similar information by experimental evolution, starting from a single gene and performing multiple cycles of in vitro mutagenesis and functional selection in Escherichia coli. We evolved two antibiotic resistance proteins, β-lactamase PSE1 and acetyltransferase AAC6, and obtained hundreds of thousands of diverse functional sequences. Using evolutionary coupling analysis, we inferred residue interaction constraints that were in agreement with contacts in known 3D structures, confirming genetic encoding of structural constraints in the selected sequences. Computational protein folding with interaction constraints then yielded 3D structures with the same fold as natural relatives. This work lays the foundation for a new experimental method (3Dseq) for protein structure determination, combining evolution experiments with inference of residue interactions from sequence information.
Links
2019
David K Yang, Samuel L Goldman, Eli Weinstein, Debora S Marks
Machine Learning in Computational Biology; Published 13 Dec 2019
Generative models for codon prediction and optimization
Abstract
Optimizing foreign DNA sequences for maximal protein production in a specified host organism is an important problem for synthetic biology and biomanufacturing. Experimental results have demonstrated that simply interchanging codons, triplets of three DNA bases, with synonymous alternatives can in fact amplify protein production several-fold while holding the produced protein constant. Previous methods for codon optimization are frequency based, which cannot consider factors such as RNA secondary structure that contribute to protein expression. Here, we apply a deep learning framework to model the distribution of codons in highly expressed bacterial and human transcripts. We show that our LSTM-Transducer model is able to predict the next codon of a genetic sequence with improved accuracy and lower perplexity on a held out set of transcripts, outperforming the previously state of the art frequency-based approach to modeling codon distribution.
Links
Anna G Green, Hadeer Elhabashy, Kelly P Brock, Rohan Maddamsetti, Oliver Kohlbacher, Debora S Marks
bioRxiv 2019; Preprint 02 Oct 2019
Proteome-scale discovery of protein interactions with residue-level resolution using sequence coevolution
Abstract
The majority of protein interactions in most organisms are unknown, and experimental methods for determining protein interactions can yield divergent results. Here we use an orthogonal, purely computational method based on sequence coevolution to discover protein interactions at large scale. In the model organism Escherichia coli, 53% of protein pairs in the proteome are eligible for our method given currently available sequenced genomes. When assaying the entire cell envelope proteome, which is understudied due to experimental challenges, we found 620 likely interactions and their predicted structures, increasing the space of known interactions by 529. Our results show that genomic sequencing data can be used to predict and resolve protein interactions to atomic resolution at large scale. Predictions and code are freely available at https://marks.hms.harvard.edu/ecolicomplex
Data availability https://marks.hms.harvard.edu/ecolicomplex
Code availability https://github.com/debbiemarkslab/EVcouplings
Links
Adam Riesselman, Jung-Eun Shin, Aaron Kollasch, Conor McMahon, Elana Simon, Chris Sander, Aashish Manglik, Andrew Kruse, Debora Marks
bioRxiv 2019; Preprint 05 Sep 2019
Accelerating Protein Design Using Autoregressive Generative Models
Abstract
A major biomedical challenge is the interpretation of genetic variation and the ability to design functional novel sequences. Since the space of all possible genetic variation is enormous, there is a concerted effort to develop reliable methods that can capture genotype to phenotype maps. State-of-art computational methods rely on models that leverage evolutionary information and capture complex interactions between residues. However, current methods are not suitable for a large number of important applications because they depend on robust protein or RNA alignments. Such applications include genetic variants with insertions and deletions, disordered proteins, and functional antibodies. Ideally, we need models that do not rely on assumptions made by multiple sequence alignments. Here we borrow from recent advances in natural language processing and speech synthesis to develop a generative deep neural network-powered autoregressive model for biological sequences that captures functional constraints without relying on an explicit alignment structure. Application to unseen experimental measurements of 42 deep mutational scans predicts the effect of insertions and deletions while matching state-of-art missense mutation prediction accuracies. We then test the model on single domain antibodies, or nanobodies, a complex target for alignment-based models due to the highly variable complementarity determining regions. We fit the model to a naïve llama immune repertoire and generate a diverse, optimized library of 105 nanobody sequences for experimental validation. Our results demonstrate the power of the ‘alignment-free’ autoregressive model in mutation effect prediction and design of traditionally challenging sequence families.
Links
John M Nicoludis, Anna G Green, Sanket Walujkar, Elizabeth J May, Marcos Sotomayor, Debora S Marks, Rachelle Gaudet
PNAS 2019; Published 20 Aug 2019
Interaction specificity of clustered protocadherins inferred from sequence covariation and structural analysis
Abstract
Clustered protocadherins, a large family of paralogous proteins that play important roles in neuronal development, provide an important case study of interaction specificity in a large eukaryotic protein family. A mammalian genome has more than 50 clustered protocadherin isoforms, which have remarkable homophilic specificity for interactions between cellular surfaces. A large antiparallel dimer interface formed by the first 4 extracellular cadherin (EC) domains controls this interaction. To understand how specificity is achieved between the numerous paralogs, we used a combination of structural and computational approaches. Molecular dynamics simulations revealed that individual EC interactions are weak and undergo binding and unbinding events, but together they form a stable complex through polyvalency. Strongly evolutionarily coupled residue pairs interacted more frequently in our simulations, suggesting that sequence coevolution can inform the frequency of interaction and biochemical nature of a residue interaction. With these simulations and sequence coevolution, we generated a statistical model of interaction energy for the clustered protocadherin family that measures the contributions of all amino acid pairs at the interface. Our interaction energy model assesses specificity for all possible pairs of isoforms, recapitulating known pairings and predicting the effects of experimental changes in isoform specificity that are consistent with literature results. Our results show that sequence coevolution can be used to understand specificity determinants in a protein family and prioritize interface amino acid substitutions to reprogram specific protein–protein interactions.
Links
Nathan J Rollins, Kelly P Brock, Frank J Poelwijk, Michael A Stiffler, Nicholas P Gauthier, Chris Sander, Debora S Marks
Nature Genetics; Published 17 June 2019
Inferring protein 3D structure from deep mutation scans
Abstract
We describe an experimental method of three-dimensional (3D) structure determination that exploits the increasing ease of high-throughput mutational scans. Inspired by the success of using natural, evolutionary sequence covariation to compute protein and RNA folds, we explored whether ‘laboratory’, synthetic sequence variation might also yield 3D structures. We analyzed five large-scale mutational scans and discovered that the pairs of residues with the largest positive epistasis in the experiments are sufficient to determine the 3D fold. We show that the strongest epistatic pairings from genetic screens of three proteins, a ribozyme and a protein interaction reveal 3D contacts within and between macromolecules. Using these experimental epistatic pairs, we compute ab initio folds for a GB1 domain (within 1.8 Å of the crystal structure) and a WW domain (2.1 Å). We propose strategies that reduce the number of mutants needed for contact prediction, suggesting that genomics-based techniques can efficiently predict 3D structure.
Links
Michael A Stiffler, Frank J Poelwijk, Kelly P Brock, Richard R Stein, Joan Teyra, Sachdev Sidhu, Debora S Marks, Nicholas P Gauthier, Chris Sander
bioRxiv 2019; Published 13 June 2019
Protein structure from experimental evolution
Abstract
Natural evolution encodes rich information about the structure and function of biomolecules in the genetic record. Previously, statistical analysis of co-variation patterns in natural protein families has enabled the accurate computation of 3D structures. Here, we explored whether similar information can be generated by laboratory evolution, starting from a single gene and performing multiple cycles of mutagenesis and functional selection. We evolved two bacterial antibiotic resistance proteins, β-lactamase PSE1 and acetyltransferase AAC6, and obtained hundreds of thousands of diverse functional sequences. Using evolutionary coupling analysis, we inferred residue interactions in good agreement with contacts in the crystal structures, confirming genetic encoding of structural constraints in the selected sequences. Computational protein folding with contact constraints yielded 3D structures with the same fold as that of natural relatives. Evolution experiments combined with inference of residue interactions from sequence information opens the door to a new experimental method for the determination of protein structures.
Links
Thomas A Hopf, Anna G Green, Benjamin Schubert, Sophia Mersmann, Charlotta PI Schärfe, John B Ingraham, Agnes Toth-Petroczy, Kelly Brock, Adam J Riesselman, Perry Palmedo, Chan Kang, Robert Sheridan, Eli J Draizen, Christian Dallago, Chris Sander, Debora S Marks
Bioinformatics
01 May 2019
The EVcouplings Python framework for coevolutionary sequence analysis
Abstract
Coevolutionary sequence analysis has become a commonly used technique for de novo prediction of the structure and function of proteins, RNA, and protein complexes. We present the EVcouplings framework, a fully integrated open-source application and Python package for coevolutionary analysis. The framework enables generation of sequence alignments, calculation and evaluation of evolutionary couplings (ECs), and de novo prediction of structure and mutation effects. The combination of an easy to use, flexible command line interface and an underlying modular Python package makes the full power of coevolutionary analyses available to entry-level and advanced users.
Links
Yuanpeng Janet Huang, Kelly P Brock, Yojiro Ishida, Gurla VT Swapna, Masayori Inouye, Debora S Marks, Chris Sander, Gaetano T Montelione
Academic Press Methods in Enzymology, pp. 363-392
Combining evolutionary covariance and NMR data for protein structure determination
Abstract
Accurate protein structure determination by solution-state NMR is challegning for proteins greater than about 20 kDa, for which extensive perdeuteration is generally required, providing experimental data that are incomplete (sparse) and ambiguous. However, the massive increase in evolutionary sequence information coupled with advanced in methods for sequence covariance analysis can provide reliable reside-residue contact information for a protein from sequence data alone. These "evolutionary couplings (ECs)" can be combined with sparse NMR data to determine accurate 3D protein structures. This hybrid "EC-NMR" method has been developed using NMR data for several soluble proteins and validated by comparison with corresponding reference structures determined by X-ray crystallography and/or conventional NMR methods. For small proteins, only backbone resonance assignments are utilized, while for larger proteins both backbone and some sidechain methyl resonance assignments are generally required. ECs can be combined with sparse NMR data obtained on deuterated, selevtively protonated protein samples to probide structures that are more accurate and complete than those obtained using such sparse NMR data alone. EC-NMR also has significant potential for analysis of protein structures from solid-state NMR data and for studies of integral membrane proteins. The requirement that ECs are consistent with NMR data recorded on a specific member of a protein family, under specific conditions, also allows identification of ECs that reflect alternative allosteric or excited states of the protein structure.
Links
Yuanpeng Janet Huang, Kelly P Brock, Chris Sander, Debora S Marks, Gaetano T Montelione
Integrative Structural Biology with Hybrid Methods; Published 08 Jan 2019
Sequence Data
Abstract
While 3D structure determination of small (< 15 kDa) proteins by solution NMR is largely automated and routine, structural analysis of larger proteins is more challenging. An emerging hybrid strategy for modeling protein structures combines sparse NMR data that can be obtained for larger proteins with sequence co-variation data, called evolutionary couplings (ECs), obtained from multiple sequence alignments of protein families. This hybrid “EC-NMR” method can be used to accurately model larger (15–60 kDa) proteins, and more rapidly determine structures of smaller (5–15 kDa) proteins using only backbone NMR data. The resulting structures have accuracies relative to reference structures comparable to those obtained with full backbone and sidechain NMR resonance assignments. The requirement that evolutionary couplings (ECs) are consistent with NMR data recorded on a specific member of a protein family, under specific conditions, potentially also allows identification of ECs that reflect alternative allosteric or excited states of the protein structure.
Links
2018
Xuewu Sui, Henning Arlt, Kelly Brock, Zon Weng Lai, Frank DiMaio, Debora Marks, Maofu Liao, Robert V Farese Jr, Tobias C Walther
Journal of Cell Biology; Published 16 October 2018
Cryo–electron microscopy structure of the lipid droplet–formation protein seipin
Abstract
Metabolic energy is stored in cells primarily as triacylglycerols in lipid droplets (LDs), and LD dysregulation leads to metabolic diseases. The formation of monolayer-bound LDs from the endoplasmic reticulum (ER) bilayer is poorly understood, but the ER protein seipin is essential to this process. In this study, we report a cryo–electron microscopy structure and functional characterization of Drosophila melanogaster seipin. The structure reveals a ring-shaped dodecamer with the luminal domain of each monomer resolved at ∼4.0 Å. Each luminal domain monomer exhibits two distinctive features: a hydrophobic helix (HH) positioned toward the ER bilayer and a β-sandwich domain with structural similarity to lipid-binding proteins. This structure and our functional testing in cells suggest a model in which seipin oligomers initially detect forming LDs in the ER via HHs and subsequently act as membrane anchors to enable lipid transfer and LD growth.
Links
Benjamin Schubert, Rohan Maddamsetti, Jackson Nyman, Maha R Farhat, Debora S Marks
Nature microbiology
03 December 2018
Genome-wide discovery of epistatic loci affecting antibiotic resistance in Neisseria gonorrhoeae using evolutionary couplings
Abstract
Genome analysis should allow the discovery of interdependent loci that together cause antibiotic resistance. In practice, however, the vast number of possible epistatic interactions erodes statistical power. Here, we extend an approach that has been successfully used to identify epistatic residues in proteins to infer genomic loci that are strongly coupled. This approach reduces the number of tests required for an epistatic genome-wide association study of antibiotic resistance and increases the likelihood of identifying causal epistasis. We discovered 38 loci and 240 epistatic pairs that influence the minimum inhibitory concentrations of 5 different antibiotics in 1,102 isolates of Neisseria gonorrhoeae that were confirmed in a second dataset of 495 isolates. Many known resistance-affecting loci were recovered; however, the majority of associations occurred in unreported genes, such as murE. About half of the discovered epistasis involved at least one locus previously associated with antibiotic resistance, including interactions between gyrA and parC. Still, many combinations involved unreported loci and genes. While most variation in minimum inhibitory concentrations could be explained by identified loci, epistasis substantially increased explained phenotypic variance. Our work provides a systematic identification of epistasis affecting antibiotic resistance in N. gonorrhoeae and a generalizable approach for epistatic genome-wide association studies.
Links
Adam J Riesselman, John B Ingraham, Debora S Marks
Nature Methods 2018; Published 24, Sep 2018
Deep generative models of genetic variation capture the effects of mutations

Abstract
The functions of proteins and RNAs are defined by the collective interactions of many residues, and yet most statistical mod-els of biological sequences consider sites nearly independently. Recent approaches have demonstrated benefits of including interactions to capture pairwise covariation, but leave higher-order dependencies out of reach. Here we show how it is pos-sible to capture higher-order, context-dependent constraints in biological sequences via latent variable models with nonlinear dependencies. We found that DeepSequence (https://github.com/debbiemarkslab/DeepSequence), a probabilistic model for sequence families, predicted the effects of mutations across a variety of deep mutational scanning experiments substantially better than existing methods based on the same evolutionary data. The model, learned in an unsupervised manner solely on the basis of sequence information, is grounded with biologically motivated priors, reveals the latent organization of sequence families, and can be used to explore new parts of sequence space.
Links
Sanduo Zheng, Lok-To Sham, Frederick A Rubino, Kelly P Brock, William P Robins, John J Mekalanos, Debora S Marks, Thomas G Bernhardt, Andrew C Kruse
PNAS 2018; Published 11 June 2018
Structure and mutagenic analysis of the lipid II flippase MurJ from Escherichia coli

Abstract
The peptidoglycan cell wall provides an essential protective barrier in almost all bacteria, defining cellular morphology and conferring resistance to osmotic stress and other environmental hazards. The precursor to peptidoglycan, lipid II, is assembled on the inner leaflet of the plasma membrane. However, peptidoglycan polymerization occurs on the outer face of the plasma membrane, and lipid II must be flipped across the membrane by the MurJ protein before its use in peptidoglycan synthesis. Due to its central role in cell wall assembly, MurJ is of fundamental importance in microbial cell biology and is a prime target for novel antibiotic development. However, relatively little is known regarding the mechanisms of MurJ function, and structural data for MurJ are available only from the extremophile Thermosipho africanus. Here, we report the crystal structure of substrate-free MurJ from the gram-negative model organism Escherichia coli, revealing an inward-open conformation. Taking advantage of the genetic tractability of E. coli, we performed high-throughput mutagenesis and next-generation sequencing to assess mutational tolerance at every amino acid in the protein, providing a detailed functional and structural map for the enzyme and identifying sites for inhibitor development. Lastly, through the use of sequence coevolution analysis, we identify functionally important interactions in the outward-open state of the protein, supporting a rocker-switch model for lipid II transport.
Links
Nathan J Rollins, Kelly P Brock, Frank J Poelwijk, Michael A Stiffler, Nicholas P Gauthier, Chris Sander, Debora S Marks
bioRxiv 2018; Published 11 May 2018
3D protein structure from genetic epistasis experiments

Abstract
High-throughput experimental techniques have made possible the systematic sampling of the single mutation landscape for many proteins, defined as the change in protein fitness as the result of point mutation sequence changes. In a more limited number of cases, and for small proteins only, we also have nearly full coverage of all possible double mutants. By comparing the phenotypic effect of two simultaneous mutations with that of the individual amino acid changes, we can evaluate epistatic effects that reflect non-additive cooperative processes. The observation that epistatic residue pairs often are in contact in the 3D structure led to the hypothesis that a systematic epistatic screen contains sufficient information to identify the 3D fold of a protein. To test this hypothesis, we examined experimental double mutants for evidence of epistasis and identified residue contacts at 86% accuracy, including secondary structure elements and evidence for an alternative all-α-helical conformation. Positively epistatic contacts – corresponding to compensatory mutations, restoring fitness – were the most informative. Folded models generated from top-ranked epistatic pairs, when compared with the known structure, were accurate within 2.4 Å over 53 residues, indicating the possibility that 3D protein folds can be determined experimentally with good accuracy from functional assays of mutant libraries, at least for small proteins. These results suggest a new experimental approach for determining protein structure.
Links
Benjamin Schubert, Charlotta Schärfe, Pierre Dönnes, Thomas Hopf, Debora S Marks, Oliver Kohlbacher
PLOS Comp Bio 2018; Published 2 March 2018
Population-specific design of de-immunized protein biotherapeutics

Abstract
Immunogenicity is a major problem during the development of biotherapeutics since it can lead to rapid clearance of the drug and adverse reactions. The challenge for biotherapeutic design is therefore to identify mutants of the protein sequence that minimize immunogenicity in a target population whilst retaining pharmaceutical activity and protein function. Current approaches are moderately successful in designing sequences with reduced immunogenicity, but do not account for the varying frequencies of different human leucocyte antigen alleles in a specific population and in addition, since many designs are non-functional, require costly experimental post-screening. Here, we report a new method for de-immunization design using multi-objective combinatorial optimization. The method simultaneously optimizes the likelihood of a functional protein sequence at the same time as minimizing its immunogenicity tailored to a target population. We bypass the need for three-dimensional protein structure or molecular simulations to identify functional designs by automatically generating sequences using probabilistic models that have been used previously for mutation effect prediction and structure prediction. As proof-of-principle we designed sequences of the C2 domain of Factor VIII and tested them experimentally, resulting in a good correlation with the predicted immunogenicity of our model.
Links
Megan Sjodt, Kelly P Brock, Genevieve Dobihal, Patricia DA Rohs, Anna G Green, Thomas A Hopf, Alexander J Meeske, Veerasak Srisuknimit, Daniel Kahne, Suzanne Walker, Debora S Marks, Thomas G Bernhardt, David Z Rudner, Andrew C Kruse
Nature 2018; Published 5 April 2018
Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis

Abstract
The shape, elongation, division and sporulation (SEDS) proteins are a large family of ubiquitous and essential transmembrane enzymes with critical roles in bacterial cell wall biology. The exact function of SEDS proteins was for a long time poorly understood, but recent work has revealed that the prototypical SEDS family member RodA is a peptidoglycan polymerase—a role previously attributed exclusively to members of the penicillin-binding protein family. This discovery has made RodA and other SEDS proteins promising targets for the development of next-generation antibiotics. However, little is known regarding the molecular basis of SEDS activity, and no structural data are available for RodA or any homologue thereof. Here we report the crystal structure of Thermus thermophilus RodA at a resolution of 2.9 Å, determined using evolutionary covariance-based fold prediction to enable molecular replacement. The structure reveals a ten-pass transmembrane fold with large extracellular loops, one of which is partially disordered. The protein contains a highly conserved cavity in the transmembrane domain, reminiscent of ligand-binding sites in transmembrane receptors. Mutagenesis experiments in Bacillus subtilis and Escherichia coli show that perturbation of this cavity abolishes RodA function both in vitro and in vivo, indicating that this cavity is catalytically essential. These results provide a framework for understanding bacterial cell wall synthesis and SEDS protein function.
Links
Benjamin Schubert, Rohan Maddamsetti, Jackson Nyman, Maha R Farhat, Debora S Marks
bioRxiv 2018; Published 1 January 2018
Genome-wide discovery of epistatic loci affecting antibiotic resistance using evolutionary couplings

Abstract
The analysis of whole genome sequencing data should, in theory, allow the discovery of interdependent loci that cause antibiotic resistance. In practice, however, identifying this epistasis remains a challenge as the vast number of possible interactions erodes statistical power. To solve this problem, we extend a method that has been successfully used to identify epistatic residues in proteins to infer loci strongly coupled and associated with antibiotic resistance from whole genomes. Our method reduces the number of tests required for an epistatic genome-wide association study and increases the likelihood of identifying causal epistasis. We discover 38 loci and 250 epistatic pairs that influence the dose needed to inhibit growth for five different antibiotics in 1102 isolates of Neisseria gonorrhoeae, that were confirmed in an independent dataset of 495 isolates. Many of the know resistance-affecting loci were recovered, and more sites within those genes, however the majority of loci occurred in unreported genes, including murE which was associated with cefixime. About half of the novel epistasis we report involves at least one locus previously associated with antibiotic resistance, including interactions between gyrA and _par_C associated with ciprofloxacin, leaving many combinations involving unreported loci and genes. Our work provides a systematic identification of epistasis pairs in N. gonorrhoeae resistance and a generalizable method for epistatic genome-wide association studies.
Links
2017
Adam J Riesselman ^, John B Ingraham ^, Debora S Marks
^ joint first authors
arXiv preprint 2017; Available on arXiv 18 December 2017
Deep Generative models of genetic variation capture mutation effects
Abstract
The functions of proteins and RNAs are determined by a myriad of interactions between their constituent residues, but most quantitative models of how molecular phenotype depends on genotype must approximate this by simple additive effects. While recent models have relaxed this constraint to also account for pairwise interactions, these approaches do not provide a tractable path towards modeling higher-order dependencies. Here, we show how latent variable models with nonlinear dependencies can be applied to capture beyond-pairwise constraints in biomolecules. We present a new probabilistic model for sequence families, DeepSequence, that can predict the effects of mutations across a variety of deep mutational scanning experiments significantly better than site independent or pairwise models that are based on the same evolutionary data. The model, learned in an unsupervised manner solely from sequence information, is grounded with biologically motivated priors, reveals latent organization of sequence families, and can be used to extrapolate to new parts of sequence space.
Links
Benjamin Schubert, Charlotta PI Schärfe, Pierre Dönnes, Thomas A Hopf, Debora S Marks, Oliver Kohlbacher
arXiv preprint 2017; Available on arXiv 28 June 2017
Population-specific design of de-immunized protein biotherapeutics
Abstract
Immunogenicity is a major problem during the development of biotherapeutics since it can lead to rapid clearance of the drug and adverse reactions. The challenge for biotherapeutic design is therefore to identify mutants of the protein sequence that minimize immunogenicity in a target population whilst retaining pharmaceutical activity and protein function. Current approaches are moderately successful in designing sequences with reduced immunogenicity, but do not account for the varying frequencies of different human leucocyte antigen alleles in a specific population and in addition, since many designs are non-functional, require costly experimental post-screening. Here we report a new method for de-immunization design using multi-objective combinatorial optimization that simultaneously optimizes the likelihood of a functional protein sequence at the same time as minimizing its immunogenicity tailored to a target population. We bypass the need for three-dimensional protein structure or molecular simulations to identify functional designs by automatically generating sequences using probabilistic models that have been used previously for mutation effect prediction and structure prediction. As proof-of-principle we designed sequences of the C2 domain of Factor VIII and tested them experimentally, resulting in a good correlation with the predicted immunogenicity of our model.
Links
John B Ingraham, Debora S Marks
ICML 2017; Available on arXiv 14 June 2017
Variational inference for sparse and undirected models

Abstract
Undirected graphical models are applied in genomics, protein structure prediction, and neuroscience to identify sparse interactions that underlie discrete data. Although Bayesian methods for inference would be favorable in these contexts, they are rarely used because they require doubly intractable Monte Carlo sampling. Here, we develop a framework for scalable Bayesian inference of discrete undirected models based on two new methods. The first is Persistent VI, an algorithm for variational inference of discrete undirected models that avoids doubly intractable MCMC and approximations of the partition function. The second is Fadeout, a reparameterization approach for variational inference under sparsity-inducing priors that captures a posteriori correlations between parameters and hyperparameters with noncentered parameterizations. We find that, together, these methods for variational inference substantially improve learning of sparse undirected graphical models in simulated and real problems from physics and biology.
Links
Charlotta PI Schärfe, Roman Tremmel, Matthias Schwab, Oliver Kohlbacher, Debora S Marks
Genome Medicine 2017; Published 22 December 2017
Genetic variation in human drug related genes

Abstract
Variability in drug efficacy and adverse effects are observed in clinical practice. While the extent of genetic variability in classic pharmacokinetic genes is rather well understood, the role of genetic variation in drug targets is typically less studied.
Links
Thomas A Hopf ^, John B Ingraham ^, Frank J Poelwijk, Charlotta PI Schärfe, Michael Springer, Chris Sander, Debora S Marks
^ Joint first authors
Nature Biotechnology; Web 16 Jan 2017
Mutation effects predicted from sequence co-variation

Abstract
Many high-throughput experimental technologies have been developed to assess the effects of large numbers of mutations (variation) on phenotypes. However, designing functional assays for these methods is challenging, and systematic testing of all combinations is impossible, so robust methods to predict the effects of genetic variation are needed. Most prediction methods exploit evolutionary sequence conservation but do not consider the interdependencies of residues or bases. We present EVmutation, an unsupervised statistical method for predicting the effects of mutations that explicitly captures residue dependencies between positions. We validate EVmutation by comparing its predictions with outcomes of high-throughput mutagenesis experiments and measurements of human disease mutations and show that it outperforms methods that do not account for epistasis. EVmutation can be used to assess the quantitative effects of mutations in genes of any organism. We provide pre-computed predictions for ∼7,000 human proteins at http://evmutation.org/.
Links
2016
Agnes Toth-Petroczy ^, Perry Palmedo ^, John B Ingraham, Thomas A Hopf, Bonnier Berger, Chris Sander, Debora S Marks
^Joint first authors
Cell, Volume 167, 158-170
Structured states of disordered proteins from genomic sequences

Abstract
Protein flexibility ranges from simple hinge movements to functional disorder. Around half of all human proteins contain apparently disordered regions with little 3D or functional information, and many of these proteins are associated with disease. Building on the evolutionary couplings approach previously successful in predicting 3D states of ordered proteins and RNA, we developed a method to predict the potential for ordered states for all apparently disordered proteins with sufficiently rich evolutionary information. The approach is highly accurate (79%) for residue interactions as tested in more than 60 known disordered regions captured in a bound or specific condition. Assessing the potential for structure of more than 1,000 apparently disordered regions of human proteins reveals a continuum of structural order with at least 50% with clear propensity for three- or two-dimensional states. Co-evolutionary constraints reveal hitherto unseen structures of functional importance in apparently disordered proteins.
Links
Yonatan H Grad, Simon R Harris, Robert D Kirkcaldy, Anna G Green, Debora S Marks, Stephen D Bentley, David Trees, Marc Lipsitch
J Infect Dis. (2016)
Genomic epidemiology of gonoccal resistance to extend Spectrum Cephalosporins, Macrolides, and Fluoroquinolones in the US, 2000-2013

Abstract
Treatment of Neisseria gonorrhoeae infection is empirical and based on population-wide susceptibilities. Increasing antimicrobial resistance underscores the potential importance of rapid diagnostic tests, including sequence-based tests, to guide therapy. However, the usefulness of sequence-based diagnostic tests depends on the prevalence and dynamics of the resistance mechanisms. We define the prevalence and dynamics of resistance markers to extended-spectrum cephalosporins, macrolides, and fluoroquinolones in 1102 resistant and susceptible clinical N. gonorrhoeae isolates collected from 2000 to 2013 via the Centers for Disease Control and Prevention's Gonococcal Isolate Surveillance Project. Reduced extended-spectrum cephalosporin susceptibility is predominantly clonal and associated with the mosaic penA XXXIV allele and derivatives (sensitivity 98% for cefixime and 91% for ceftriaxone), but alternative resistance mechanisms have sporadically emerged. Reduced azithromycin susceptibility has arisen through multiple mechanisms and shows limited clonal spread; the basis for resistance in 36% of isolates with reduced azithromycin susceptibility is unclear. Quinolone-resistant N. gonorrhoeae has arisen multiple times, with extensive clonal spread. Quinolone-resistant N. gonorrhoeae and reduced cefixime susceptibility appear amenable to development of sequence-based diagnostic tests, whereas the undefined mechanisms of resistance to ceftriaxone and azithromycin underscore the importance of phenotypic surveillance. The identification of multidrug-resistant isolates highlights the need for additional measures to respond to the threat of untreatable gonorrhea.
Links
John M Nicoludis, Bennett E Vogt, Anna G Green, Charlotta PI Schärfe, Debora S marks, Rachelle Gaudet
elife; 2016;5:e18449
Antiparallel protocadherin homodimers use distinct affinity and specificity-mediating regions in cadherin repeats 1-4
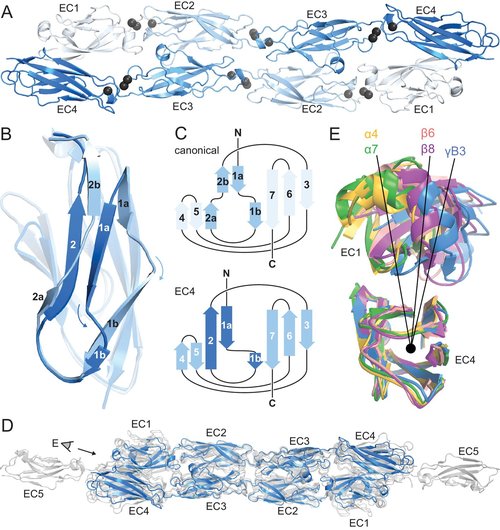
Abstract
Protocadherins (Pcdhs) are cell adhesion and signaling proteins used by neurons to develop and maintain neuronal networks, relying on trans homophilic interactions between their extracellular cadherin (EC) repeat domains. We present the structure of the antiparallel EC1-4 homodimer of human PcdhγB3, a member of the γ subfamily of clustered Pcdhs. Structure and sequence comparisons of α, β, and γ clustered Pcdh isoforms illustrate that subfamilies encode specificity in distinct ways through diversification of loop region structure and composition in EC2 and EC3, which contains isoform-specific conservation of primarily polar residues. In contrast, the EC1/EC4 interface comprises hydrophobic interactions that provide non-selective dimerization affinity. Using sequence coevolution analysis, we found evidence for a similar antiparallel EC1-4 interaction in non-clustered Pcdh families. We thus deduce that the EC1-4 antiparallel homodimer is a general interaction strategy that evolved before the divergence of these distinct protocadherin families.
Links
Caleb Weinrab, Adam J Riesselman, John B Ingraham, Torsten GRoss, Chris Sander, Debora S Marks
Cell, Volume 165, 1-13
3D RNA and functional interactions from evolutionary couplings

Abstract
Non-coding RNAs are ubiquitous, but the discovery of new RNA gene sequences far outpaces the research on the structure and functional interactions of these RNA gene sequences. We mine the evolutionary sequence record to derive precise information about the function and structure of RNAs and RNA-protein complexes. As in protein structure prediction, we use maximum entropy global probability models of sequence co-variation to infer evolutionarily constrained nucleotide-nucleotide interactions within RNA molecules and nucleotide-amino acid interactions in RNA-protein complexes. The predicted contacts allow all-atom blinded 3D structure prediction at good accuracy for several known RNA structures and RNA-protein complexes. For unknown structures, we predict contacts in 160 non-coding RNA families. Beyond 3D structure prediction, evolutionary couplings help identify important functional interactions—e.g., at switch points in riboswitches and at a complex nucleation site in HIV. Aided by increasing sequence accumulation, evolutionary coupling analysis can accelerate the discovery of functional interactions and 3D structures involving RNA.
Links
2015
John M Nicoludis, Sze-i Lau, Charlotta PI Schärfe, Debora S Marks, Wilhelm A Weihofen, Rachelle Gaudet
Cell Press Vol.23 no. 11 pp. 2087-2098
Structure and sequence analyses of clustered protocadherins reveal antiparallel interactions that mediate homophilic specificity

Abstract
Clustered protocadherin (Pcdh) proteins mediate dendritic self-avoidance in neurons via specific homophilic interactions in their extracellular cadherin (EC) domains. We determined crystal structures of EC1–EC3, containing the homophilic specificity-determining region, of two mouse clustered Pcdh isoforms (PcdhγA1 and PcdhγC3) to investigate the nature of the homophilic interaction. Within the crystal lattices, we observe antiparallel interfaces consistent with a role in trans cell-cell contact. Antiparallel dimerization is supported by evolutionary correlations. Two interfaces, located primarily on EC2-EC3, involve distinctive clustered Pcdh structure and sequence motifs, lack predicted glycosylation sites, and contain residues highly conserved in orthologs but not paralogs, pointing toward their biological significance as homophilic interaction interfaces. These two interfaces are similar yet distinct, reflecting a possible difference in interaction architecture between clustered Pcdh subfamilies. These structures initiate a molecular understanding of clustered Pcdh assemblies that are required to produce functional neuronal networks.
Links
Thomas A Hopf ^, John B Ingraham ^, Frank J Poelwijk, Michael Springer, Chris Sander, Debora S Marks
^Joint first authors
Quantification of the effects of mutations using a global probability model of natural sequence variation
Abstract
Modern biomedicine is challenged to predict the effects of genetic variation. Systematic functional assays of point mutants of proteins have provided valuable empirical information, but vast regions of sequence space remain unexplored. Fortunately, the mutation-selection process of natural evolution has recorded rich information in the diversity of natural protein sequences. Here, building on probabilistic models for correlated amino-acid substitutions that have been successfully applied to determine the three-dimensional structures of proteins, we present a statistical approach for quantifying the contribution of residues and their interactions to protein function, using a statistical energy, the evolutionary Hamiltonian. We find that these probability models predict the experimental effects of mutations with reasonable accuracy for a number of proteins, especially where the selective pressure is similar to the evolutionary pressure on the protein, such as antibiotics.
Links
Yuefeng Tang, Yuanpeng Janet Huang, Thomas A Hopf, Chris Sander ^, Debora S Marks ^, Gaetano T Montelione ^
^Corresponding authors
Nature Methods 12, 751-754
Protein Structure determinationby combining sparse NMR data with evolutionary couplings

Abstract
Accurate determination of protein structure by NMR spectroscopy is challenging for larger proteins, for which experimental data are often incomplete and ambiguous. Evolutionary sequence information together with advances in maximum entropy statistical methods provide a rich complementary source of structural constraints. We have developed a hybrid approach (evolutionary coupling–NMR spectroscopy; EC-NMR) combining sparse NMR data with evolutionary residue-residue couplings and demonstrate accurate structure determination for several proteins 6−41 kDa in size.
Links
Richard R Stein, Debora S Marks, Chris Sander
PLoS Comput Biol 11(7): e1004182
Inferring pairwise interactions from biological data using maximum-entropy probability models
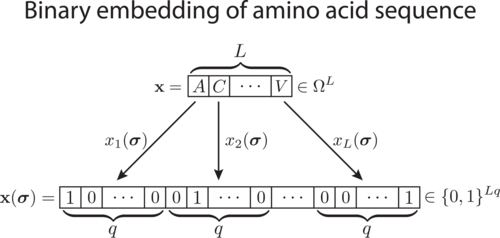
Abstract
Maximum entropy-based inference methods have been successfully used to infer direct interactions from biological datasets such as gene expression data or sequence ensembles. Here, we review undirected pairwise maximum-entropy probability models in two categories of data types, those with continuous and categorical random variables. As a concrete example, we present recently developed inference methods from the field of protein contact prediction and show that a basic set of assumptions leads to similar solution strategies for inferring the model parameters in both variable types. These parameters reflect interactive couplings between observables, which can be used to predict global properties of the biological system. Such methods are applicable to the important problems of protein 3-D structure prediction and association of gene–gene networks, and they enable potential applications to the analysis of gene alteration patterns and to protein design.
Links
Sikander Hayat, Chris Sander, Debora S Marks, Arne Elofsson
PNAS Vol. 112 no. 17 pp. 5413-5418
All-atom 3D structure prediction of transmembrane β-barrel proteins from sequences

Abstract
Transmembrane β-barrels (TMBs) carry out major functions in substrate transport and protein biogenesis but experimental determination of their 3D structure is challenging. Encouraged by successful de novo 3D structure prediction of globular and α-helical membrane proteins from sequence alignments alone, we developed an approach to predict the 3D structure of TMBs. The approach combines the maximum-entropy evolutionary coupling method for predicting residue contacts (EVfold) with a machine-learning approach (boctopus2) for predicting β-strands in the barrel. In a blinded test for 19 TMB proteins of known structure that have a sufficient number of diverse homologous sequences available, this combined method (EVfold_bb) predicts hydrogen-bonded residue pairs between adjacent β-strands at an accuracy of ∼70%. This accuracy is sufficient for the generation of all-atom 3D models. In the transmembrane barrel region, the average 3D structure accuracy [template-modeling (TM) score] of top-ranked models is 0.54 (ranging from 0.36 to 0.85), with a higher (44%) number of residue pairs in correct strand–strand registration than in earlier methods (18%). Although the nonbarrel regions are predicted less accurately overall, the evolutionary couplings identify some highly constrained loop residues and, for FecA protein, the barrel including the structure of a plug domain can be accurately modeled (TM score = 0.68). Lower prediction accuracy tends to be associated with insufficient sequence information and we therefore expect increasing numbers of β-barrel families to become accessible to accurate 3D structure prediction as the number of available sequences increases.
Links
Jörn M Schmiedel, Sandy L Klemm, Yannan Zheng, Apratim Sahay, Nils Blüthgen, Debora S marks, Alexander van Oudenaarden
Science Vol. 348 no. 6230 pp. 128-132
MicroRNA control of protein expression noise
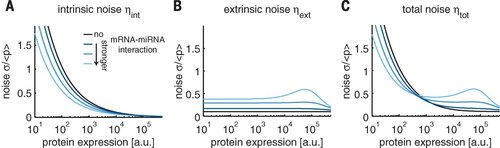
Abstract
MicroRNAs (miRNAs) repress the expression of many genes in metazoans by accelerating messenger RNA degradation and inhibiting translation, thereby reducing the level of protein. However, miRNAs only slightly reduce the mean expression of most targeted proteins, leading to speculation about their role in the variability, or noise, of protein expression. We used mathematical modeling and single-cell reporter assays to show that miRNAs, in conjunction with increased transcription, decrease protein expression noise for lowly expressed genes but increase noise for highly expressed genes. Genes that are regulated by multiple miRNAs show more-pronounced noise reduction. We estimate that hundreds of (lowly expressed) genes in mouse embryonic stem cells have reduced noise due to substantial miRNA regulation. Our findings suggest that miRNAs confer precision to protein expression and thus offer plausible explanations for the commonly observed combinatorial targeting of endogenous genes by multiple miRNAs, as well as the preferential targeting of lowly expressed genes.
Links
Robert Sheridan, Robert J Fieldhouse, Sikander Hayat, Yichao Sun, Yevgeniy Antipin, Li Yang, Thomas A Hopf, Debora S Marks, Chris Sander
Evolutionary Couplings and protein 3D structure prediction

Abstract
Recently developed maximum entropy methods infer evolutionary constraints on protein function and structure from the millions of protein sequences available in genomic databases. The EVfold web server (at EVfold.org) makes these methods available to predict functional and structural interactions in proteins. The key algorithmic development has been to disentangle direct and indirect residue-residue correlations in large multiple sequence alignments and derive direct residue-residue evolutionary couplings (EVcouplings or ECs). For proteins of unknown structure, distance constraints obtained from evolutionarily couplings between residue pairs are used to de novo predict all-atom 3D structures, often to good accuracy. Given sufficient sequence information in a protein family, this is a major advance toward solving the problem of computing the native 3D fold of proteins from sequence information alone.
Links
2014
Thomas A Hopf, Satoshi Morinaga, Sayoko Ihara, Kazushige Touhara, Debora S Marks, Richard Benton
Nature Communications 6, Article number: 6077
Amino acid coevolution reveals three-dimensional strucutre and functional domains of insect odorant receptors

Abstract
Insect odorant receptors (ORs) comprise an enormous protein family that translates environmental chemical signals into neuronal electrical activity. These heptahelical receptors are proposed to function as ligand-gated ion channels and/or to act metabotropically as G protein-coupled receptors (GPCRs). Resolving their signalling mechanism has been hampered by the lack of tertiary structural information and primary sequence similarity to other proteins. We use amino acid evolutionary covariation across these ORs to define restraints on structural proximity of residue pairs, which permit de novo generation of three-dimensional models. The validity of our analysis is supported by the location of functionally important residues in highly constrained regions of the protein. Importantly, insect OR models exhibit a distinct transmembrane domain packing arrangement to that of canonical GPCRs, establishing the structural unrelatedness of these receptor families. The evolutionary couplings and models predict odour binding and ion conduction domains, and provide a template for rationale structure-activity dissection.
Links
Thomas A Hopf ^, Charlotta PI Schärfe, João PGLM Rodrigues, Anna G Green, Oliver Kohlbacher, Chris Sander, Alexandre MJJ Bonvin, Debora S Marks
^Joint first authors
elife 2014;3:e03430
Sequence co-evolution gives 3D contacts and structures of protein complexes

Abstract
Protein–protein interactions are fundamental to many biological processes. Experimental screens have identified tens of thousands of interactions, and structural biology has provided detailed functional insight for select 3D protein complexes. An alternative rich source of information about protein interactions is the evolutionary sequence record. Building on earlier work, we show that analysis of correlated evolutionary sequence changes across proteins identifies residues that are close in space with sufficient accuracy to determine the three-dimensional structure of the protein complexes. We evaluate prediction performance in blinded tests on 76 complexes of known 3D structure, predict protein–protein contacts in 32 complexes of unknown structure, and demonstrate how evolutionary couplings can be used to distinguish between interacting and non-interacting protein pairs in a large complex. With the current growth of sequences, we expect that the method can be generalized to genome-wide elucidation of protein–protein interaction networks and used for interaction predictions at residue resolution.
Links
2012
Debora S Marks, Thomas A Hopf, Chris Sander
Nature Biotechnology 30, pp. 1072-1080(2012)
Protein structure prediction from sequence variation

Abstract
Genomic sequences contain rich evolutionary information about functional constraints on macromolecules such as proteins. This information can be efficiently mined to detect evolutionary couplings between residues in proteins and address the long-standing challenge to compute protein three-dimensional structures from amino acid sequences. Substantial progress has recently been made on this problem owing to the explosive growth in available sequences and the application of global statistical methods. In addition to three-dimensional structure, the improved understanding of covariation may help identify functional residues involved in ligand binding, protein-complex formation and conformational changes. We expect computation of covariation patterns to complement experimental structural biology in elucidating the full spectrum of protein structures, their functional interactions and evolutionary dynamics.
Links
Thomas A Hopf, Lucy J Colwell, Robert Sheridan, Burkhard Rost, Chris Sander, Debora S Marks
Cell, Vol. 149, Issue 7, pp. 1707-1721
Three-Dimensional structure of membrane proteins from genomic sequencing

Abstract
We show that amino acid covariation in proteins, extracted from the evolutionary sequence record, can be used to fold transmembrane proteins. We use this technique to predict previously unknown 3D structures for 11 transmembrane proteins (with up to 14 helices) from their sequences alone. The prediction method (EVfold_membrane) applies a maximum entropy approach to infer evolutionary covariation in pairs of sequence positions within a protein family and then generates all-atom models with the derived pairwise distance constraints. We benchmark the approach with blinded de novo computation of known transmembrane protein structures from 23 families, demonstrating unprecedented accuracy of the method for large transmembrane proteins. We show how the method can predict oligomerization, functional sites, and conformational changes in transmembrane proteins. With the rapid rise in large-scale sequencing, more accurate and more comprehensive information on evolutionary constraints can be decoded from genetic variation, greatly expanding the repertoire of transmembrane proteins amenable to modeling by this method.
Links
2011 and earlier
Debora S Marks ^, Lucy J Colwell ^, Robert Sheridan, Thomas A Hopf, Andrea Pagnani, Riccardo Zecchina, Chris Sander
^Joint first authors
PLoS One 2011 June 12 :e28766. Epub* 2011 Dec 7
Protein 3D structure computed from evolutionary sequence variation
Abstract
The evolutionary trajectory of a protein through sequence space is constrained by its function. Collections of sequence homologs record the outcomes of millions of evolutionary experiments in which the protein evolves according to these constraints. Deciphering the evolutionary record held in these sequences and exploiting it for predictive and engineering purposes presents a formidable challenge. The potential benefit of solving this challenge is amplified by the advent of inexpensive high-throughput genomic sequencing.
In this paper we ask whether we can infer evolutionary constraints from a set of sequence homologs of a protein. The challenge is to distinguish true co-evolution couplings from the noisy set of observed correlations. We address this challenge using a maximum entropy model of the protein sequence, constrained by the statistics of the multiple sequence alignment, to infer residue pair couplings. Surprisingly, we find that the strength of these inferred couplings is an excellent predictor of residue-residue proximity in folded structures. Indeed, the top-scoring residue couplings are sufficiently accurate and well-distributed to define the 3D protein fold with remarkable accuracy.
We quantify this observation by computing, from sequence alone, all-atom 3D structures of fifteen test proteins from different fold classes, ranging in size from 50 to 260 residues., including a G-protein coupled receptor. These blinded inferences are de novo, i.e., they do not use homology modeling or sequence-similar fragments from known structures. The co-evolution signals provide sufficient information to determine accurate 3D protein structure to 2.7–4.8 Å Cα-RMSD error relative to the observed structure, over at least two-thirds of the protein (method called EVfold, details at http://EVfold.org). This discovery provides insight into essential interactions constraining protein evolution and will facilitate a comprehensive survey of the universe of protein structures, new strategies in protein and drug design, and the identification of functional genetic variants in normal and disease genomes.
Links
Aaron Arvey, Erik Larsson, Chris Sander, Christina S Leslie, Debora S Marks
Molecular Systems Biology 6 (2010): 363. PMC. Web. 13 Oct. 2016
Target mRNA abundance dilutes microRNA and siRNA activity

Abstract
Post-transcriptional regulation by microRNAs and siRNAs depends not only on characteristics of individual binding sites in target mRNA molecules, but also on system-level properties such as overall molecular concentrations. We hypothesize that an intracellular pool of microRNAs/siRNAs faced with a larger number of available predicted target transcripts will downregulate each individual target gene to a lesser extent. To test this hypothesis, we analyzed mRNA expression change from 178 microRNA and siRNA transfection experiments in two cell lines. We find that downregulation of particular genes mediated by microRNAs and siRNAs indeed varies with the total concentration of available target transcripts. We conclude that to interpret and design experiments involving gene regulation by small RNAs, global properties, such as target mRNA abundance, need to be considered in addition to local determinants. We propose that analysis of microRNA/siRNA targeting would benefit from a more quantitative definition, rather than simple categorization of genes as 'target' or 'not a target.' Our results are important for understanding microRNA regulation and may also have implications for siRNA design and small RNA therapeutics.
Links
Aly A Khan, Doron Betel, Martin L Miller, Chris Sander, Christina S Leslie, Debora S Marks
Nature Biotechnology, 2009:27(6):549-55
Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs
![]()
Abstract
Transfection of small RNAs (such as small interfering RNAs (siRNAs) and microRNAs (miRNAs)) into cells typically lowers expression of many genes. Unexpectedly, increased expression of genes also occurs. We investigated whether this upregulation results from a saturation effect—that is, competition among the transfected small RNAs and the endogenous pool of miRNAs for the intracellular machinery that processes small RNAs. To test this hypothesis, we analyzed genome-wide transcript responses from 151 published transfection experiments in seven different human cell types. We show that targets of endogenous miRNAs are expressed at significantly higher levels after transfection, consistent with impaired effectiveness of endogenous miRNA repression. This effect exhibited concentration and temporal dependence. Notably, the profile of endogenous miRNAs can be largely inferred by correlating miRNA sites with gene expression changes after transfections. The competition and saturation effects have practical implications for miRNA target prediction, the design of siRNA and short hairpin RNA (shRNA) genomic screens and siRNA therapeutics.
Links
Bino John, Anton J Enright, Alexei Aravin, Thomas Tuschl, Chris Sander, Debora S Marks
PLoS Biology :e363. Epub 2004
Human microRNA targets

Abstract
MicroRNAs (miRNAs) interact with target mRNAs at specific sites to induce cleavage of the message or inhibit translation. The specific function of most mammalian miRNAs is unknown. We have predicted target sites on the 3′ untranslated regions of human gene transcripts for all currently known 218 mammalian miRNAs to facilitate focused experiments. We report about 2,000 human genes with miRNA target sites conserved in mammals and about 250 human genes conserved as targets between mammals and fish. The prediction algorithm optimizes sequence complementarity using position-specific rules and relies on strict requirements of interspecies conservation. Experimental support for the validity of the method comes from known targets and from strong enrichment of predicted targets in mRNAs associated with the fragile X mental retardation protein in mammals. This is consistent with the hypothesis that miRNAs act as sequence-specific adaptors in the interaction of ribonuclear particles with translationally regulated messages. Overrepresented groups of targets include mRNAs coding for transcription factors, components of the miRNA machinery, and other proteins involved in translational regulation, as well as components of the ubiquitin machinery, representing novel feedback loops in gene regulation. Detailed information about target genes, target processes, and open-source software for target prediction (miRanda) is available at http://www.microrna.org. Our analysis suggests that miRNA genes, which are about 1% of all human genes, regulate protein production for 10% or more of all human genes.